Troubleshooting
Chronic Disease Films
Chronic Disease Films 2
M. tuberculosis genomes
M. tuberculosis PacBio reads
100
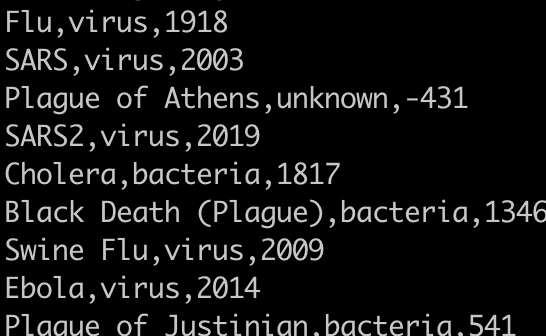
How many instances of the flu are there?
grep -c "flu" pandemics.csv
This will give you a count of zero because grep is case sensitive. You need to either:
1) use the -i flag to ignore case or
2) capitalize the first f in flu.
100
How many movies are there?
Hint: There is a header line.
# Subtract one for the header line or grep it out
wc -l chronic_disease_movies.txt
cut -f 1 chronic_disease_movies.txt | wc -l
awk -F "\t" chronic_disease_movies.txt | wc -l
100
How many movies were released in 2017?
grep -c 2017 chronic_disease_movies.txt
grep 2017 chronic_disease_movies.txt|wc -l
100
How many sequences are there in myctu.fa?
Note: M. tuberculosis has only one chromosome.
grep -c '>' myctu.fa
or
bioawk -c fastx '{print seq}' myctu.fa | wc -l
100
How many rows are there in a fastq file for each sequence?
4
200
Log onto the server
ssh jschmoe@inbre.ncgr.org
This is missing the port (2406).
ssh -p 2406 jschmoe@inbre.ncgr.org
200
Print only the year (column 2).
cut -f 2 chronic_disease_movies.txt
awk -F "\t" '{print $2}' chronic_disease_movies.txt
200
Which movie is about Lady Gaga?
grep "Lady Gaga" chronic_disease_movies.txt
200
M. tuberculosis has one chromosome but there are more genome sequences in myctu.fa than the number of M. tuberculosis strains with their genomes sequenced. Why?
Some assemblies are fragmented (the chromosome was assembled into multiple pieces).
200
How many sequence reads are there in M_tub_SRR17234886.fastq.gz?
bioawk -c fastx '{print $seq}' M_tub_SRR17234886.fastq.gz | wc -l
300
Link to a file.
ln -s myfile.txt
It needs a destination. You can put in a new file name or just use "." to name it the same thing.
ln -s myfile.txt newfile.txt
or
ln -s myfile.txt .
300
Print only the movie column (1st column).
cut -f 1 chronic_disease_movies.txt
awk -F "\t" '{print $1}' chronic_disease_movies.txt
300
Sort the file by year (second column)
sort -t $'\t' -nk2 chronic_disease_movies.txt
300
How many genome sequences represent a complete genome?
Note: they will have "complete sequence" in the header.
grep -c "complete sequence" myctu.fa
or
awk '$1~/complete sequence/{print}' myctu.fa | wc -l
300
To get a sense for the lengths of these sequence reads, find how long are first 10 sequences are in M_tub_SRR17234886.fastq.gz.
bioawk -c fastx '{print length($seq)}' M_tub_SRR17234886.fastq.gz | head
400
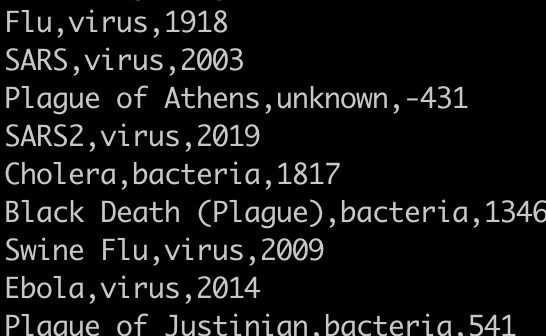
Print the pathogen column
awk '{print $2}' pandemics.csv
1) You need to change the delimiter to a comma.
2) Alternatively, you could use:
cut -d, -f2 pandemics.csv
400
Grab all lines with movies made before 2000 (column 2)
awk -F "\t" '$2<2000{print}' chronic_disease_movies.txt
awk -F "\t" '$2<=1999{print}' chronic_disease_movies.txt
400
Which movies have "Cancer" in the "Disease" column (column 3)?
awk -F "\t" '$3=="Cancer" {print}' chronic_disease_movies.txt
400
How long are the first 10 sequences in myctu.fa?
bioawk -c fastx '{print length($seq)}' myctu.fa
400
Count the number of sequence reads >= 10,000 nts in M_tub_SRR17234886.fastq.gz.
bioawk -c fastx 'length($seq)>=10000{print}' M_tub_SRR17234886.fastq.gz | wc -l
or
bioawk -c fastx '{print $name}' M_tub_SRR17234886.fastq.gz | sed "s/length=//" | awk '$1>=10000{print}' | wc -l
500
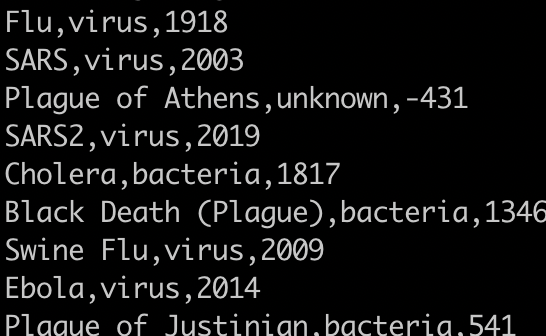
Find the unique pathogens.
awk -F, '{print $2}' sort.pandemics.csv | uniq
You need to sort it first. If you run it like this it will only collapse duplicates that are on adjacent rows. You will get: virus, unknown, virus, bacteria, virus, bacteria.
1) awk -F, '{print $2}' sort.pandemics.csv | sort | uniq
1) awk -F, '{print $2}' sort.pandemics.csv | sort -u
500
Grab all lines with movies that were made between 2000 and 2010 (column 2).
awk -F "\t" '$2>=2000&&<=2010{print}' chronic_disease_movies.txt
etc
500
How many movies were made in each year?
# You can also grep -v Year if you want to remove the header line
cut -f 2 chronic_disease_movies.txt | sort | uniq -c
awk -F "\t" '{print $2}' chronic_disease_movies.txt |sort | uniq -c
500
How long are the 10 longest genome sequences in myctu.fa?
bioawk -c fastx '{print length($seq)}' myctu.fa | sort -n | tail
or
bioawk -c fastx '{print length($seq)}' myctu.fa | sort -nr | head
500
What are the longest and shortest read lengths in M_tub_SRR17234886.fastq.gz?
bioawk -c fastx '{print length($seq)}' M_tub_SRR17234886.fastq.gz | wc -l
or
bioawk -c fastx '{print $name}' M_tub_SRR17234886.fastq.gz | sed "s/length=//" | awk '$1>=10000{print}' | wc -l