Genetic Disorders Pt. 1
Genetic Disorders Pt. 2
Genetic Disorders Pt. 3
Metabolism Pt. 1
Metabolism Pt. 2
100
Associated with short stature, broad webbed neck, L sided congenital heart defects like bicuspid aortic valve or coarctation of the aorta.
Infants may present with lymphedema of hands and feet.
Turner Syndrome (Primary Gonadal Dysgenesis)
100
May present as a teenager who is tall and infertile with small testes and minimal intellectual disability.
Klinefelter syndrome (47 XXY)
100
Previously known as Von Recklinghausen Disease. Autosomal dominant on chromosome 17. Children with this disorder can get pheochromocytoma and renal artery stenosis and need to be monitored for hypertension.
Neurofibromatosis Type 1
100
Infants with this disorder will appear normal at birth until they have lactose for the first time then they will present with poor feeding and failure to thrive. If they are not treated then they can get cataracts, intellectual disability and liver disease.
Galactosemia
Due to deficiency of galactose-1-phosphate uridyltransferase
100
Patients with this disease are asymptomatic for a few months before presenting with severe vomiting, irritability, eczema and a musty or mousy odor of the urine. This disease is now easily identified on newborn screening.
Phenylketonuria (PKU)
200
Classic features include hypertelorism, unusually-shaped chest, webbed neck, low set ears, and pulmonic stenosis despite normal karyotype.
Noonan Syndrome
200
Classic features include neonatal hypotonia, failure to thrive as an infant with voracious appetite starting around 2-3 years of age and leading to obesity.
Prader-Willi Syndrome
200
Children with this syndrome have elfin facies with wide spaced teeth and upturned nose, hypercalcemia, supravalvular aortic stenosis and a "cocktail party personality."
Williams Syndrome
200
This disease is due to a deficiency of hepatic glucose-6-phosphatase. Infants have a doll-like face. They will likely present once they are sleeping through the night which results in prolonged fasting and manifests as hypoglycemia and lactic acidosis.
Glycogen Storage Disease Type 1 (Von Gierke Disease)
200
This syndrome presents before age 2 with progressive facial coarsening, hirsutism, thick skulls, corneal clouding and severe intellectual disability.
Hurler Syndrome (Mucopolysaccharidoses Type 1)
300
Usually an autosomal dominant connective tissue disorder associated with skeletal, cardiac, and ophthalmologic ramifications. The mutation is on chromosome 15 in the fibrillar gene.
Marfan Syndrome
300
Children with this syndrome have behavioral anomalies such as frequent laughter, easy excitability, or short attention span. They usually have microcephaly, seizures, spasticity, and feeding problems.
Angelman Syndrome
300
You need a chromosomal microarray to diagnose this contiguous gene syndrome that involves a micro deletion and an autosomal dominant inheritance pattern. Affected children may have tetralogy of fallout, thymic aplasia, hypoparathyroidism and cleft palate.
22q11 deletion syndrome or Velocardiofacial Syndrome
Formerly DiGeorge Syndrome
300
This disease is due to a deficiency in lysosomal breakdown of glycogen. Infants are normal at birth, but become floppy by 1 month of age though their muscles are hard on exam. They have cardiomegaly, hepatomegaly, and macroglossia.
Glycogen Storage Disease Type II (Pompe Disease)
300
Patients with this syndrome have coarse facial features in addition to being short with skeletal abnormalities. It is X linked recessive.
Hunter syndrome (MPS Type II)
400
Children with this syndrome are at increased risk for leukemia, duodenal atresia, AV canal (endocardial cushion defect) and atlantoaxial instability.
Trisomy 21 (Down Syndrome)
400
This overgrowth syndrome due to an alteration in Chromosome 11 p15.5 puts children at risk for embryonal tumors like rhabdomyosarcoma, Wilm's tumor (nephroblastoma) and neuroblastoma.
Beckwith Weidemann Syndrome
400
One of the most common causes of combined deafness and blindness. This syndrome also has heart and GU abnormalities with delayed growth and development.
CHARGE syndrome
Coloboma
Heart Defects
Atresia Choanae
Retardation of Growth/Development
GU Abnormalities
Ear anomalies
400
This disease onsets during the first week of life. Infants have tachypnea, shallow breathing, and encephalopathy. Infants have elevated plasma levels of valine, isoleucine, alloisoleucine, and leucine by the end of the first day of life. Lastly, their urine has a classic smell that isn't so bad.
Maple Syrup Urine Disease
400
Children with this lipid storage disease present with hepatosplenomegaly, bone pain, short stature and easy bruisability.
Gaucher Disease
500
Features include rocker bottom feet, clenched fist with overlapping fingers, hypoplastic nails, prominent occiput, microcephaly, microophthalmia, low set malformed ears, horseshoe kidney, and seizures. There are severe growth and developmental delays.
Trisomy 18
500
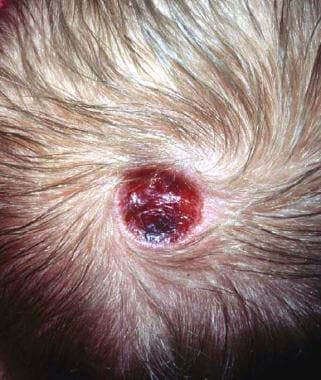
The trisomy associated with this exam finding.
Trisomy 13
Cutis aplasia (punched-out scalp lesions) is the exam finding.
500
Classic features of this syndrome are broad thumbs and cryptorchidism.
Rubenstein-Taybi Syndrome
500
This condition presents with a body type similar to Marfan's but the lens is usually displaced downwards instead of the upwards displacement seen with Marfan's. It is also associated with intellectual disability and thrombosis. There is an error in methionine metabolism.
Homocystinuria
500
Patients with this disease have normal development through the first 9 months and then start to have lethargy and hypotonia. They are owed to have exaggerated startle reflex, cherry red spot on the retina, and macrocephaly on exam. Unlike Niemann-Pick Disease, there is no hepatosplenomegaly.
Tay Sachs Disease