Clinical Cases
Enzyme Errors
Local Lens
Labs & Imaging
Integration
100
A previously healthy 8-month-old boy is brought to the emergency department due to lethargy and poor feeding. Physical examination reveals hepatomegaly. Laboratory studies show severe fasting hypoglycemia, elevated serum lactate, hypertriglyceridemia, and hyperuricemia. Urine ketones are present. The patient’s symptoms improve with frequent carbohydrate-rich feedings. Which of the following enzyme deficiencies best explains this patient’s condition?
A) Debranching enzyme
B) Glycogen phosphorylase
C) Acid alpha-glucosidase
D) Glucose-6-phosphatase
E) Muscle phosphorylase
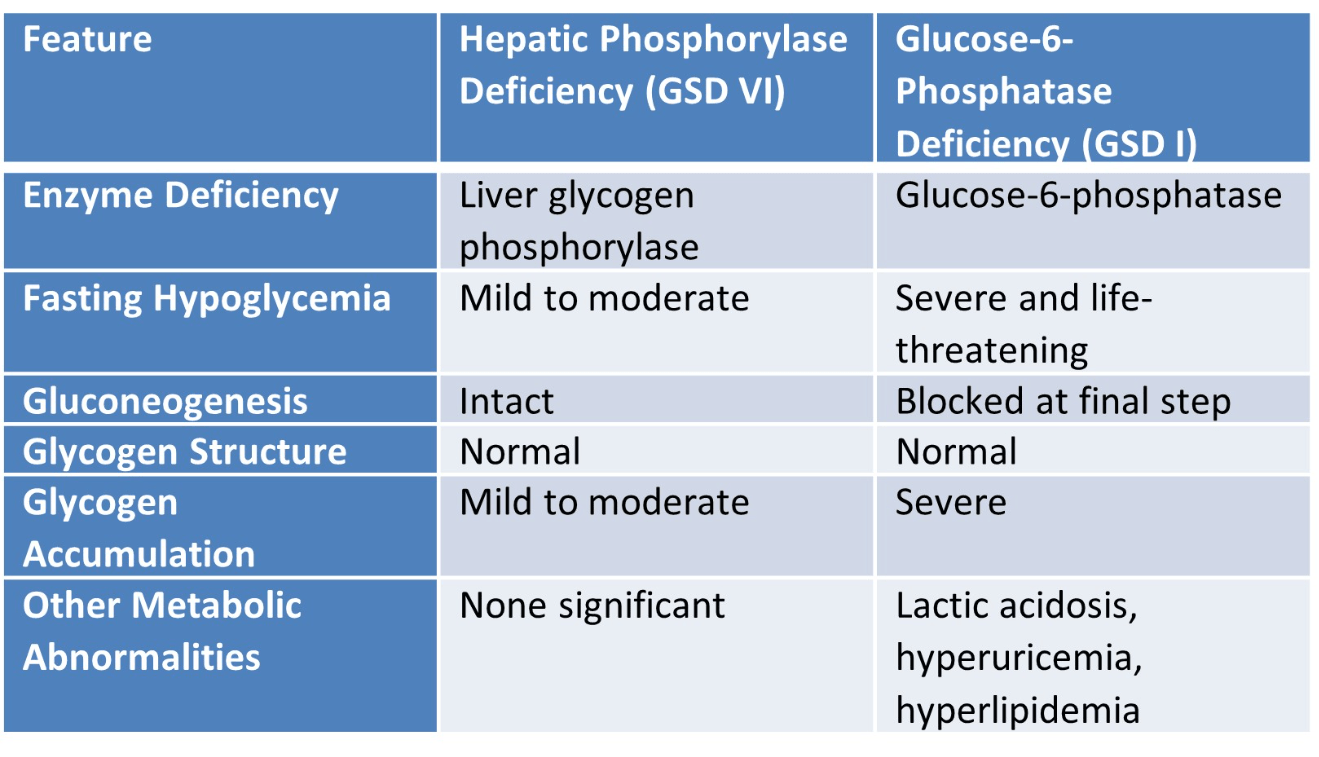
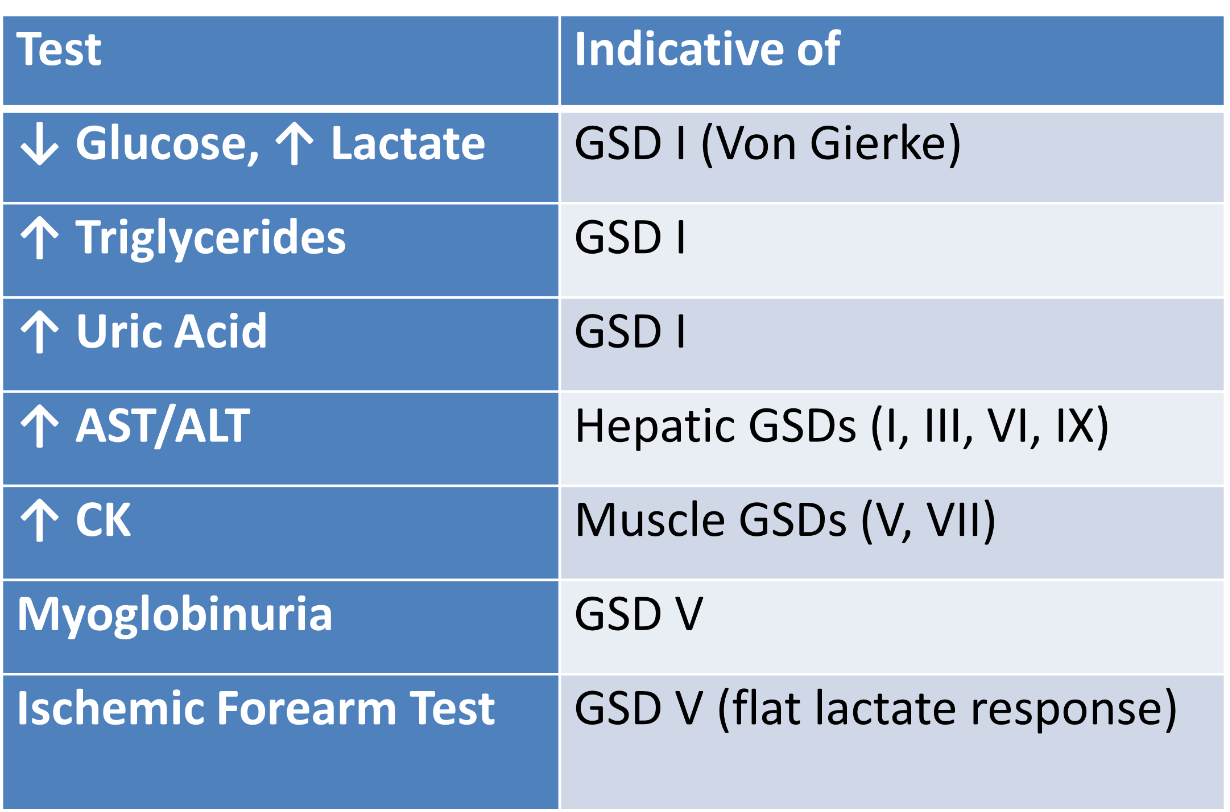
This clinical scenario is characteristic of Glycogen Storage Disease Type I (Von Gierke disease), which is caused by a deficiency in glucose-6-phosphatase. This enzyme is required for the final step of both glycogenolysis and gluconeogenesis, converting glucose-6-phosphate into free glucose. Without it, glucose cannot be released into the bloodstream, resulting in severe fasting hypoglycemia, lactic acidosis, hyperuricemia, and hyperlipidemia. Hepatomegaly occurs due to glycogen accumulation in the liver. The patient's improvement with frequent feedings reflects the reliance on exogenous glucose in the absence of adequate endogenous glucose production.
100
A 4-month-old infant is brought to the pediatrician for evaluation of feeding difficulties, poor weight gain, and progressive weakness. On examination, the infant has macroglossia, hypotonia, and hepatomegaly. Cardiac auscultation reveals a gallop rhythm. Chest X-ray shows cardiomegaly. Serum creatine kinase is elevated. Which of the following additional findings is most likely to be present in this patient, and what is the underlying enzyme deficiency?
A) Hypoglycemia due to glucose-6-phosphatase deficiency
B) Hepatic fibrosis due to abnormal glycogen branching
C) Lysosomal glycogen accumulation due to acid alpha-glucosidase deficiency
D) Muscle cramps and myoglobinuria due to phosphorylase deficiency
E) Hepatomegaly with hyperlipidemia due to debranching enzyme deficiency
The presentation is classic for Pompe disease (GSD Type II)—an autosomal recessive disorder caused by deficiency of acid alpha-glucosidase (GAA), a lysosomal enzyme. This leads to accumulation of glycogen primarily in cardiac, skeletal, and smooth muscle.
Typical findings include:
Cardiomegaly and hypertrophic cardiomyopathy
Hypotonia (floppy baby)
Macroglossia
Elevated CK
Normal blood glucose (distinguishes it from hepatic GSDs)
Pompe disease does not typically cause fasting hypoglycemia, as glycogenolysis in the cytoplasm is intact. The key distinguishing feature is lysosomal glycogen buildup, affecting muscle function and cardiac integrity.
200
A 3-year-old child is brought to the clinic for evaluation of hepatomegaly and growth delay. The parents report that the child becomes unusually tired and irritable when meals are delayed. Physical examination reveals a protuberant abdomen and mild hypotonia. Laboratory tests reveal mild fasting hypoglycemia and elevated liver transaminases. A liver biopsy shows excessive accumulation of glycogen with abnormally short outer chains. Which of the following enzyme deficiencies is most likely responsible for this patient's findings, and what intermediate is expected to accumulate in affected tissues?
A) Glycogen synthase deficiency; glucose-1-phosphate
B) Debranching enzyme deficiency; limit dextrin
C) Glucose-6-phosphatase deficiency; glucose-6-phosphate
D) Branching enzyme deficiency; amylopectin-like glycogen
E) Muscle phosphorylase deficiency; lactate
Debranching enzyme deficiency; limit dextrin
This patient presents with signs consistent with Glycogen Storage Disease Type III (Cori disease), due to deficiency of the debranching enzyme (amylo-1,6-glucosidase). This enzyme is responsible for removing branch points in glycogen during degradation. Its absence leads to the accumulation of limit dextrin, a partially degraded glycogen molecule with short outer chains, seen on liver biopsy.
Compared to GSD I, Cori disease typically presents with milder fasting hypoglycemia because gluconeogenesis remains functional. Hepatomegaly and growth delay result from glycogen accumulation and chronic energy deficiency.
200
A newborn male of Ashkenazi Jewish descent is brought to the clinic for evaluation of abdominal distension. Physical examination reveals hepatomegaly. The parents report that an older sibling had similar symptoms and was later diagnosed with a metabolic liver disorder. Initial labs reveal mild fasting hypoglycemia and elevated triglycerides. Genetic testing is being considered. Which of the following glycogen storage disorders should be prioritized in the differential diagnosis, and why?
A) GSD II and GSD IV, because they are associated with abnormal lysosomal glycogen degradation in this population
B) GSD I and GSD III, due to higher carrier frequency of pathogenic variants in this ancestry
C) GSD V, because it is the most common autosomal recessive muscle GSD worldwide
D) GSD VI, because it is the most benign form and doesn’t require genetic testing
E) GSD I and GSD IX, due to X-linked inheritance and neonatal onset
B) GSD I and GSD III, due to higher carrier frequency of pathogenic variants in this ancestry
Explanation:
The Ashkenazi Jewish population has a higher carrier frequency of mutations in genes associated with Glycogen Storage Disease Type I (G6PC gene) and Type III (AGL gene). These conditions often present with hepatomegaly, fasting hypoglycemia, and hyperlipidemia in infancy or early childhood.
GSD I (Von Gierke disease) is particularly common in this group due to founder mutations. Recognizing the patient's ancestry allows for targeted genetic testing, which can expedite diagnosis and management.
Other options are incorrect:
GSD II and IV involve different pathologies (lysosomal vs. branching enzyme) and are not specifically associated with Ashkenazi ancestry.
GSD V primarily affects muscle, not liver.
GSD VI and IX are less severe, and GSD IX is X-linked, but less strongly associated with this ancestry.
200
A 2-year-old boy is evaluated for hepatomegaly and mild fasting hypoglycemia. Physical examination reveals a protuberant abdomen but no signs of muscle weakness or developmental delay. Laboratory studies show a low fasting glucose that significantly increases following intramuscular administration of glucagon. Which of the following enzymes is most likely functioning normally in this patient?
A) Glucose-6-phosphatase
B) Acid alpha-glucosidase
C) Muscle glycogen phosphorylase
D) Liver glycogen phosphorylase
E) Debranching enzyme
Glucagon stimulates glycogenolysis via activation of liver glycogen phosphorylase, which breaks down glycogen into glucose-1-phosphate (and eventually glucose). The rise in glucose following glucagon administration indicates that hepatic glycogenolysis is intact, and liver phosphorylase is functional.
This finding helps differentiate between GSD types:
In GSD I (glucose-6-phosphatase deficiency), glucose levels do not rise after glucagon, because glucose-6-phosphate cannot be converted to free glucose.
In GSD VI (Hers disease), liver phosphorylase is deficient, and glucagon has minimal effect.
Acid alpha-glucosidase (Pompe disease) affects lysosomal degradation, not acute glucose regulation.
Muscle glycogen phosphorylase is not relevant to hepatic glucose response.
400
Compare hepatic phosphorylase and glucose-6-phosphatase deficiencies regarding fasting tolerance and glycogen structure.
400
A rural pediatrician lacks access to genetic panels. What biochemical tests aid GSD diagnosis and referral?
400
A 1-year-old girl is evaluated for progressive abdominal distension and poor weight gain. Physical examination reveals hepatomegaly and signs of early liver dysfunction. A liver biopsy shows extensive fibrosis with abnormal glycogen deposits that are poorly branched and stain similarly to amylopectin. Genetic testing confirms a deficiency in the glycogen branching enzyme. Which of the following best explains the progression to cirrhosis in this patient, and how does this mechanism differ from other glycogen storage diseases?
A) Excessive accumulation of normally structured glycogen triggers inflammation and hepatocyte apoptosis
B) Impaired glycogen breakdown leads to severe fasting hypoglycemia and secondary liver injury
C) Abnormal glycogen structure triggers chronic inflammation and fibrosis, unlike the non-toxic glycogen accumulation in other GSDs
D) Inability to release glucose leads to hepatomegaly and hyperlipidemia but not liver fibrosis
E) Lysosomal accumulation of glycogen impairs mitochondrial function and leads to cirrhosis
This child has Glycogen Storage Disease Type IV (Andersen disease), caused by deficiency of the glycogen branching enzyme (GBE1). The resulting glycogen has fewer branch points, forming amylopectin-like, poorly soluble polymers that accumulate in the liver. These abnormal glycogen structures are cytotoxic, inducing inflammation, fibrosis, and eventually cirrhosis.
In contrast, most other GSDs (e.g., GSD I, III, VI) involve the accumulation of structurally normal glycogen, which is non-inflammatory and typically causes metabolic issues (like hypoglycemia) but not progressive liver damage.
Other choices:
A) describes normal glycogen accumulation, which doesn’t trigger inflammation.
B) applies to GSD I but doesn’t cause fibrosis.
D) is more consistent with GSD I or III, which are not fibrogenic.
E) refers to Pompe disease (GSD II), which affects muscle, not liver fibrosis.
500
A previously healthy 8-year-old boy is evaluated for recurrent episodes of muscle pain and dark-colored urine after physical activity. These symptoms occur particularly after strenuous exercise, such as running. Laboratory testing shows elevated serum creatine kinase and myoglobinuria. An ischemic forearm exercise test shows no significant rise in serum lactate. Which of the following is the most appropriate set of lifestyle and dietary recommendations for this patient?
A) High-protein diet and resistance training
B) Ketogenic diet and unrestricted physical activity
C) Pre-exercise glucose supplementation and low-intensity aerobic conditioning
D) High-fat diet and avoidance of all physical activity
E) High-fiber diet and frequent rest periods during exercise
Pre-exercise carbohydrate intake, avoid anaerobic exertion, moderate aerobic training
Explanation: Muscle phosphorylase deficiency limits ATP generation; exogenous glucose helps energy supply, while aerobic conditioning uses fat metabolism.
500
A 2-year-old girl is evaluated for hepatomegaly and growth delay. Her parents report that she becomes irritable and lethargic when meals are delayed. Physical examination reveals a protuberant abdomen and mild hypotonia. Fasting laboratory studies reveal mild hypoglycemia with elevated liver transaminases. A liver biopsy shows glycogen with abnormally short outer branches and accumulation within hepatocytes.
Which of the following enzyme deficiencies is most likely responsible for this patient's condition, and what is the best method to confirm the diagnosis?
A) Glucose-6-phosphatase deficiency; confirm with serum lactate and uric acid levels
B) Debranching enzyme deficiency; confirm with enzyme assay or AGL gene sequencing
C) Branching enzyme deficiency; confirm with muscle biopsy
D) Liver phosphorylase deficiency; confirm with ischemic forearm exercise test
E) Acid alpha-glucosidase deficiency; confirm with cardiac MRI
This patient’s clinical and histologic findings are characteristic of Glycogen Storage Disease Type III (Cori or Forbes disease), caused by a deficiency in the debranching enzyme (amylo-1,6-glucosidase). The hallmark is accumulation of glycogen with short outer chains (limit dextrins), seen on liver biopsy, along with fasting intolerance, hepatomegaly, and growth delay.
The diagnosis can be confirmed by:
Enzyme assay (from liver, muscle, or leukocytes)
Genetic testing of the AGL gene, which encodes the debranching enzyme
Other options are incorrect:
G6Pase deficiency (GSD I) shows normal glycogen structure but with severe hypoglycemia and lactic acidosis.
Branching enzyme deficiency (GSD IV) produces amylopectin-like deposits and leads to early cirrhosis.
Liver phosphorylase deficiency (GSD VI) shows normal glycogen structure.
Acid alpha-glucosidase deficiency (Pompe disease) affects muscle and heart, not primarily the liver.