The Irony
HIT Me Up!
Thrombo-cytowhat?
Bloody Brilliant
Mash Up
100
Approximately 85% to 90% of patients with hereditary hemochromatosis are homozygous for what gene mutation?
A:C282Y
B:H63D
C:S65C
D:TP53
Approximately 85% to 90% of patients with HH are homozygous for the C282Y mutation in the HFE gene; the remaining patients are typically compound heterozygotes for C282Y and the H63D or S65C mutations.
100
What is the mechanism of action of Heparin?

Heparin binds to antithrombin III which results in a conformational change which increases its affinity for factor Xa and factor IIa (thrombin) there by inactivating each factor. This in turn decreases the conversion of prothrombin to thrombin and fibrinogen to fibrin respectively ultimately resulting in decreased fibrin clot formation
100
Immune thrombocytopenic purpura (ITP) is defined as isolated thrombocytopenia (platelet count <100,000/microL) without anemia or leukopenia and without another apparent cause of the thrombocytopenia. It is caused by antibodies against glycoproteins on the cell surface of platelets leading to platelet destruction. While most patients with ITP are asymptomatic, those with symptoms may present with petechiae and ecchymosis without concurrent lymphadenopathy or splenomegaly. More severe cases may present with continued epistaxis and even severe hemorrhage. Laboratory evaluation is often limited to isolated thrombocytopenia and peripheral smear will often show the presence of large platelets but no evidence of other abnormal cells such as schistocytes.
While ITP is a diagnosis of exclusion, several potential triggers exist which should be screened for based on findings from the history and physical exam. That said, all patients with ITP should be tested for what two specific infections per MKSAP?

100
What is the first step in hemostasis?
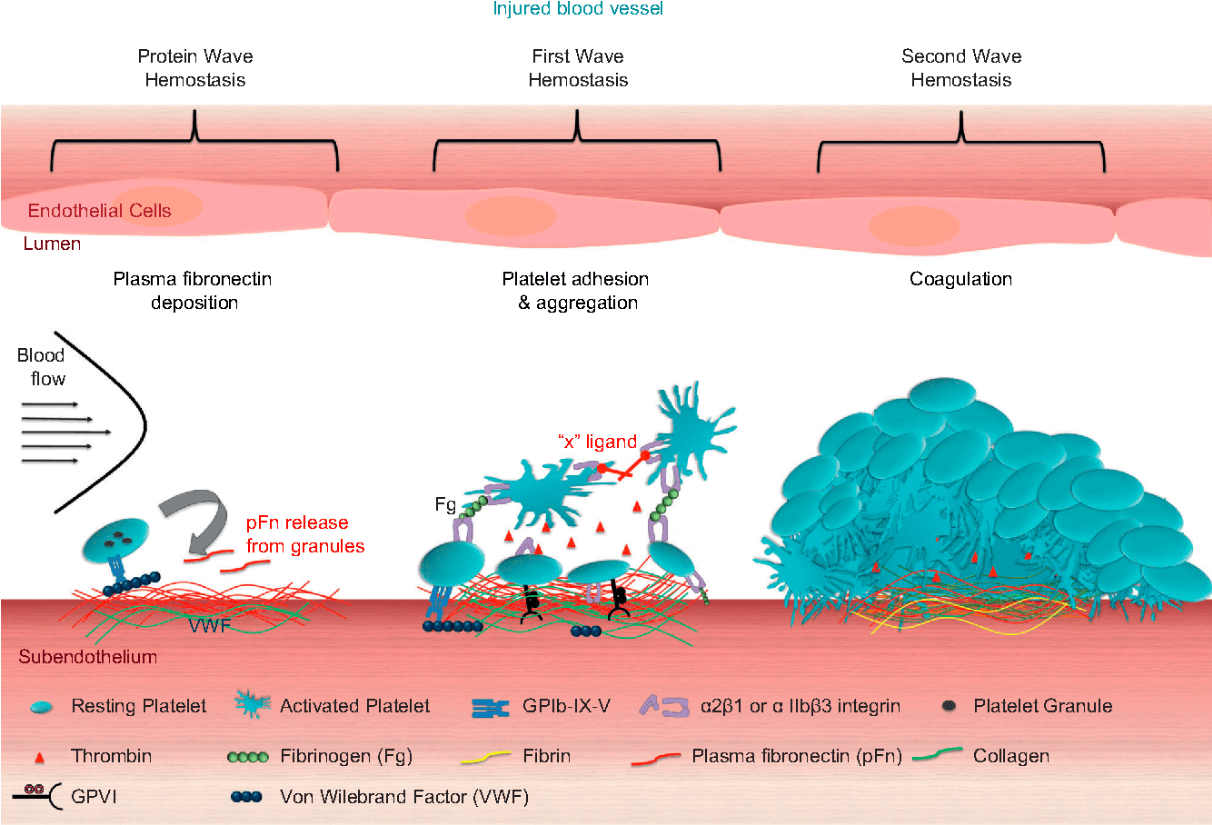
Platelet activation is the first step in hemostasis which occurs in a three-stage process.
Platelets adhere to exposed subendothelial surfaces of injured vessel walls (mediated by von Willebrand factor [vWF]).
Platelets then change shape and release adenosine diphosphate and triphosphate, serotonin, fibrinogen, vWF, and factor V. During shape change, glycoprotein IIb/IIIa receptors are exposed, bind fibrinogen, and initiate reversible platelet-platelet aggregation.
In the final stage, arachidonic acid is released and converted to thromboxane A2, promoting irreversible platelet aggregation and resulting in a platelet plug.
100
What platlet count level is required for surgical intervention in most individuals?
Although 100,000/µL is desirable for neurosurgery, platelet counts between 30,000 and 50,000/µL must be corrected before surgical intervention, and counts less than 10,000/µL are associated with risk of spontaneous bleeding.
200
How is hereditary hemochromatosis diagnosed?
What two organs are most affected?
The diagnosis is suspected by finding an elevated transferrin saturation greater than 45% and an elevated serum ferritin level, characteristic arthropathy, endocrinopathy such as androgen insufficiency, and/or skin hyper-pigmentation "bronze diabetes"; diagnosis is confirmed through genetic testing. First-degree relatives of patients with hemochromatosis should be screened.
LIVER AND HEART:
Cirrhosis and cardiomyopathy are major causes of morbidity; patients may be diagnosed late in the disease course through evaluation of decompensated liver disease or symptomatic cardiomyopathy, although liver and heart damage are largely irreversible at this late stage even if iron stores are actively reduced.
200
Describe the pathogenesis of HIT type II and explain the mechanisms that lead to thrombocytopenia and thrombosis
 The primary pathogenesis of HIT type II is the formation of HIT antibodies (IgG antibodies against platelet factor 4 (PF4) complexed to heparin). The antigen/antibody complex further binds to the Fc receptor on the cell surface of platelets and endothelial cells.
The primary pathogenesis of HIT type II is the formation of HIT antibodies (IgG antibodies against platelet factor 4 (PF4) complexed to heparin). The antigen/antibody complex further binds to the Fc receptor on the cell surface of platelets and endothelial cells.
Thrombosis: Binding to the Fc receptor of the surface of platelets results in platelet activation resulting in the release of procoagulant substances including additional PF4. This ultimately leads to increased platelet activation and aggregation. Likewise, binding to Fc receptors on endothelial cells leads to increased tissue factor expression and thrombin generation as well as increase in release of adhesion molecules like IL-6 and von Willebrand factor ultimately resulting in a prothrombotic state.
Thrombocytopenia: Platelets coated in IgG are removed from circulation by splenic macrophages. Platelets are also consumed by inappropriate thrombosis as detailed above
200
In patients with ITP, under what conditions is outpatient therapy appropriate and what initial therapy should be offered to said patients?
It is recommended to initiate outpatient therapy in asymptomatic patients or those with minor mucocutaneous bleeding who present with platelet counts between 20,000/uL and 30,000/uL. MKSAP recommends initial treatment with a short course (<6 weeks) of dexamethasone or prednisone. UpToDate further elaborates this as oral dexamethasone, 40mg daily for 4 days with no taper or oral prednisone 1mg/kg daily for 1-2 weeks followed by gradual taper (with a preference for the dexamethasone treatment option).
200
What should be the first step in evaluation of isolated thrombocytopenia?
The first step of evaluation is to review the peripheral smear to confirm the count. Pseudothrombocytopenia may occur in patients with antibodies to ethylenediaminetetraacetic acid (EDTA). an accurate count can be obtained from blood drawn in citrate or heparin instead of EDTA.
200
Who long should antithrombotic therapy be continued in patients with HIT?
In patients without document thromboembolic events, anticoagulation should be continued for 2-3 months.
In patients with document thromboembolic events, anticoagulation should be continued for 3-6 months.
Patients should be instructed to avoid heparin for life.
300
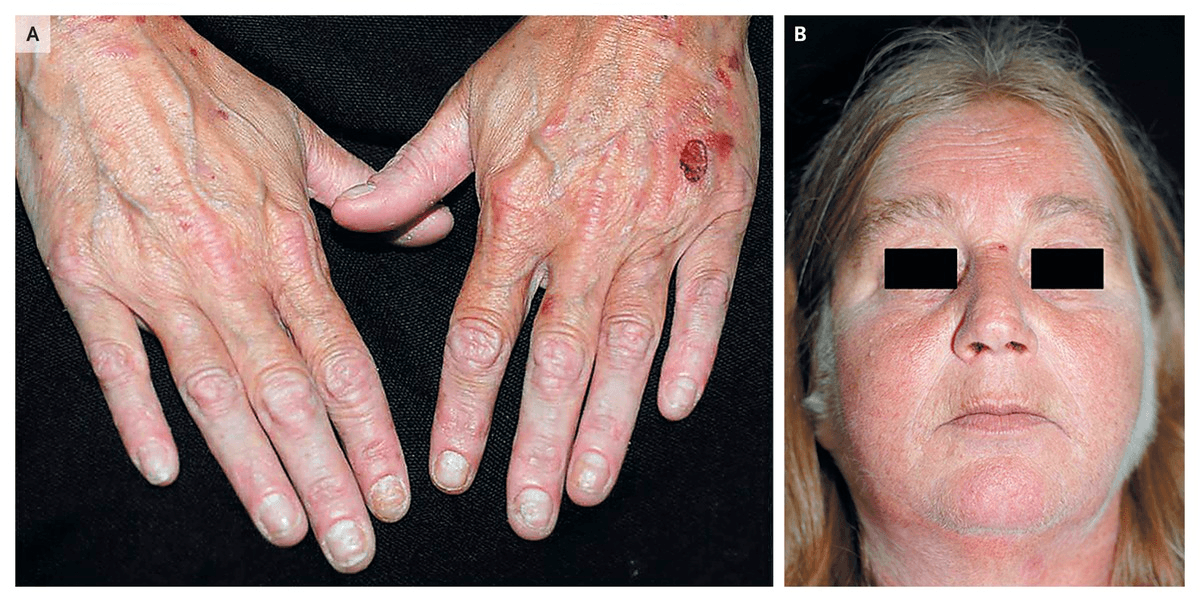
What disease process has these two characteristic findings present?
Bonus: How do you treat it?
Porphyria cutanea tarda is characterized by cutaneous blisters, often on the hands, and hypertrichosis
Treatment? It responds well to phlebotomy.
300
What are the four categories of the 4T Scoring System for predicting Heparin-Induced Thrombocytopenia and Thrombosis? and how many points to do you need to qualify for each of the different probability categories?
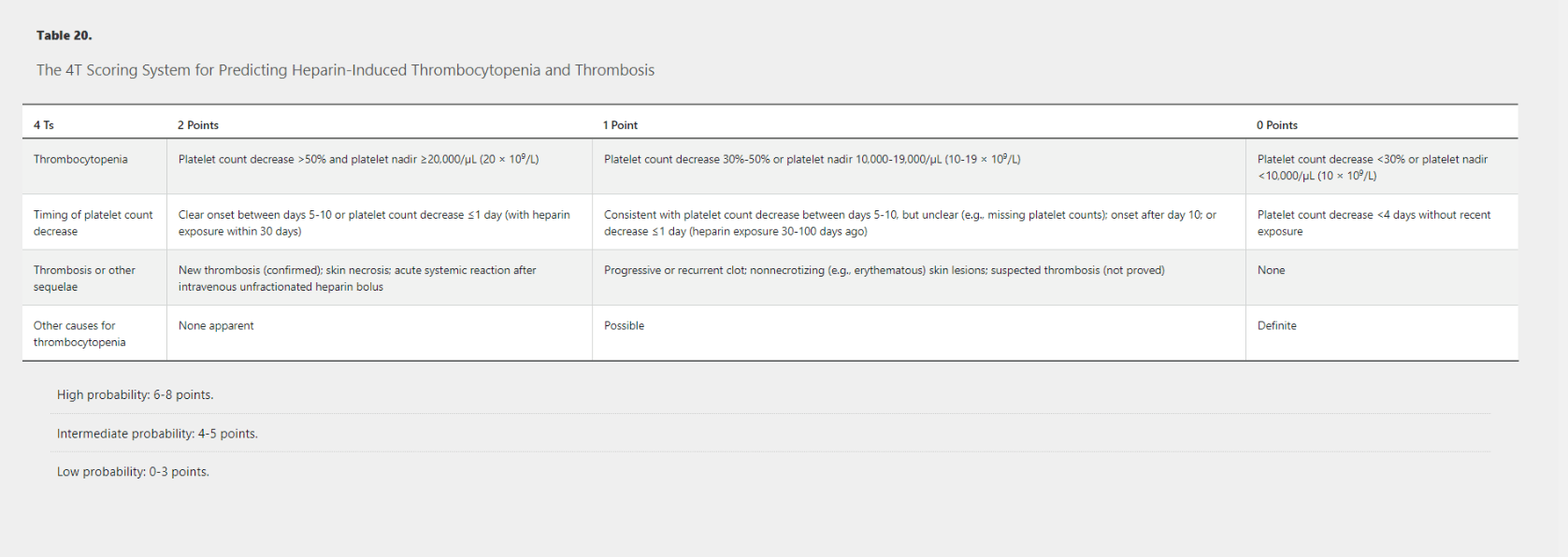
300
Hemolytic Uremic Syndrome presents as a triade of thrombocytopenia, microangiopathic hemolytic anemia, and acute kidney injury. It classically presents following diarrheal illness caused by EHEC O157:H7 and is ultimately caused by direct damage to endothelial cells by released shiga-like toxin resulting in inappropriately prothrombotic states in small vessels, including those found within the kidney. This leads to inappropriate thrombosis resulting in platelet sequestration as well as red cell lysis. Antibiotics are not recommended in patients with classic HUS as they do not change the disease course or may even worsen the severity of HUS due to increased toxin release from killed bacteria.
Classic HUS has a 4% associated mortality and patients often suffer from long-term sequelae of hypertension and mild kidney disease. While treatment for non-severe cases is typically supportive, how should severe cases be addressed?
The mainstay of HUS treatment is supportive care with a focus on fluid resuscitation. Blood transfusions are indicated if Hgb drops below 7 or in patients with significantly symptomatic anemia. Platelet transfusions are often deferred as they may worsen the disease process but may be appropriate if patients have severe thrombocytopenia (<20,000/uL) and overt bleeding or in patients with thrombocytopenia (<50,000/uL) and require major invasive procedures. Dialysis is used following its usual indications. Plasma exchange may also be appropriate in patients with severe HUS or in those whom the distinction from TTP is unclear.
300
How is classic Hemolytic Uremic Syndrome (HUS) diagnosed? What is it's management?
Diagnosis is confirmed by stool culture and polymerase chain reaction for Shiga toxin, free Shiga toxin, or enterotoxin-producing E. coli O157:H7 antigen testing.
Management is supportive for most patients; antibiotics do not alter the natural course of disease.
300
What distinguishes hypertrichosis from hirsutism?
Hypertrichosis is excessive hair growth anywhere on the body in either males or females. (It has also been called werewolf syndrome as hair may grow up to 10 centimeters in length, giving a "werewolf" appearance)
Hirsutism, which is a term reserved for females who grow an excessive amount of terminal hairs in androgen-dependent sites
400
A 76-year-old woman is evaluated for worsening fatigue. She reports no additional symptoms. Medical history is significant for MDS diagnosed 3 years ago, which has required the transfusion of 1 to 2 units of blood every 5 to 6 weeks to maintain a hemoglobin level of approximately 8 g/dL (80 g/L); she has exertional dyspnea when the hemoglobin level drops below 7 g/dL (70 g/L).
On physical examination, vital signs are normal. The abdomen is soft. Conjunctival pallor is noted. The examination is otherwise noncontributory.
Laboratory studies show a hemoglobin level of 7.5 g/dL (75 g/L) and a serum ferritin level of 1638 ng/mL (1638 µg/L).
Which of the following is the most appropriate management?
A: Perform therapeutic phlebotomy
B: Begin deferasirox
C: Begin succimer
D: Wait until hemoglobin drops below 7, then transfuse
B: Begin deferasirox
In most cases, secondary iron overload occurs in patients with severe anemia who require chronic transfusion therapy.
Therapeutic phlebotomy is contraindicated in most patients with secondary iron overload because they are already anemic and often still transfusion dependent.
Treat with iron chelation with an oral drug, such as deferasirox or deferiprone.
400
When HIT is suspected, at what point should Heparin products be discontinued? How would you formally confirm the diagnosis of HIT.

Heparin products should be discontinued if the 4T score is greater than or equal to 4.
Per MSKAP 18, confirmation of HIT is a 2 step process:
1. Sensitive screening test (negative result rules out the diagnosis): enzyme-linked immunosorbent assay for PF4 antibodies
2. Confirmatory test: functional test measuring either serotonin release assay or the heparin-induced platelet aggregation assay
400
Describe the pathogenesis of Thrombotic Thrombocytopenic Purpura (TTP)
TTP is caused by a deficiency in metalloprotease ADAMTS13 which is responsible for cleaving high-molecular weight vWF multimers. This leads to aggregation of the multimers resulting in platelet rich thrombi in small vessels (causing platelet consumption and thrombocytopenia) and in turn causes shear stress on erythrocytes (resulting in MAHA and presence of schistocytes on peripheral smear).
Deficiency in metalloprotease ADAMTS13 can be caused by natural deficiency caused by genetic mutation (hereditary TTP), or more commonly development of antibodies against ADAMTS13. Drugs such as ticlopidine, quinine, cyclosporine, gemcitabine, and vascular endothelial growth factor inhibitors (such as bevacizumab) can cause TTP. Drug abuse with oxymorphone, 3,4-methylenedioxymethamphetamine (“ecstasy”), and cocaine has also been reported to cause TTP.
400
Thrombotic thrombocytopenic purpura (TTP) presents with thrombocytopenia and microangiopathic hemolytic anemia (MAHA); other clinical features may include fever, kidney disease, and fluctuating neurologic abnormalities.
What are the causes?
Autoantibodies to ADAMTS13 (cleaves the high-molecular-weight multimers of von Willebrand factor (vWF)) are responsible for the deficiency in most patients, but hereditary TTP has been reported in some.
Drugs such as ticlopidine, quinine, cyclosporine, gemcitabine, and vascular endothelial growth factor inhibitors (such as bevacizumab) can cause TTP. Drug abuse with oxymorphone, 3,4-methylenedioxymethamphetamine (“ecstasy”), and cocaine has also been reported to cause TTP.
400
A 68-year-old man is evaluated for epistaxis, gum bleeding, and easy bruising of 3 months' duration. Medical history is notable for anxiety, depression, and hyperlipidemia. Medications are atorvastatin, citalopram, a multivitamin, and ginkgo biloba.
On physical examination, vital signs are normal; BMI is 21. Scattered petechiae and several small ecchymoses are visible on the anterior thigh. The examination is otherwise normal.
Labs:
Hemoglobin: 14.8 g/dL (148 g/L)
Leukocyte count: 4200/µL (4.2 × 109/L)
Platelet count: 245,000/µL (245 × 109/L)
PT, aPTT, and thrombin time: normal
What is the most appropriate diagnostic test to perform next?
- Patients presenting with mucocutaneous bleeding and normal complete blood count, prothrombin time, and activated partial thromboplastin time should be evaluated for von Willebrand disease or acquired platelet dysfunction.
- The Platelet Function Analyzer-100 is a useful screening test for assessing qualitative platelet disorders. It is a sensitive screen for von Willebrand disease and will detect abnormalities caused by antiplatelet medications.
500
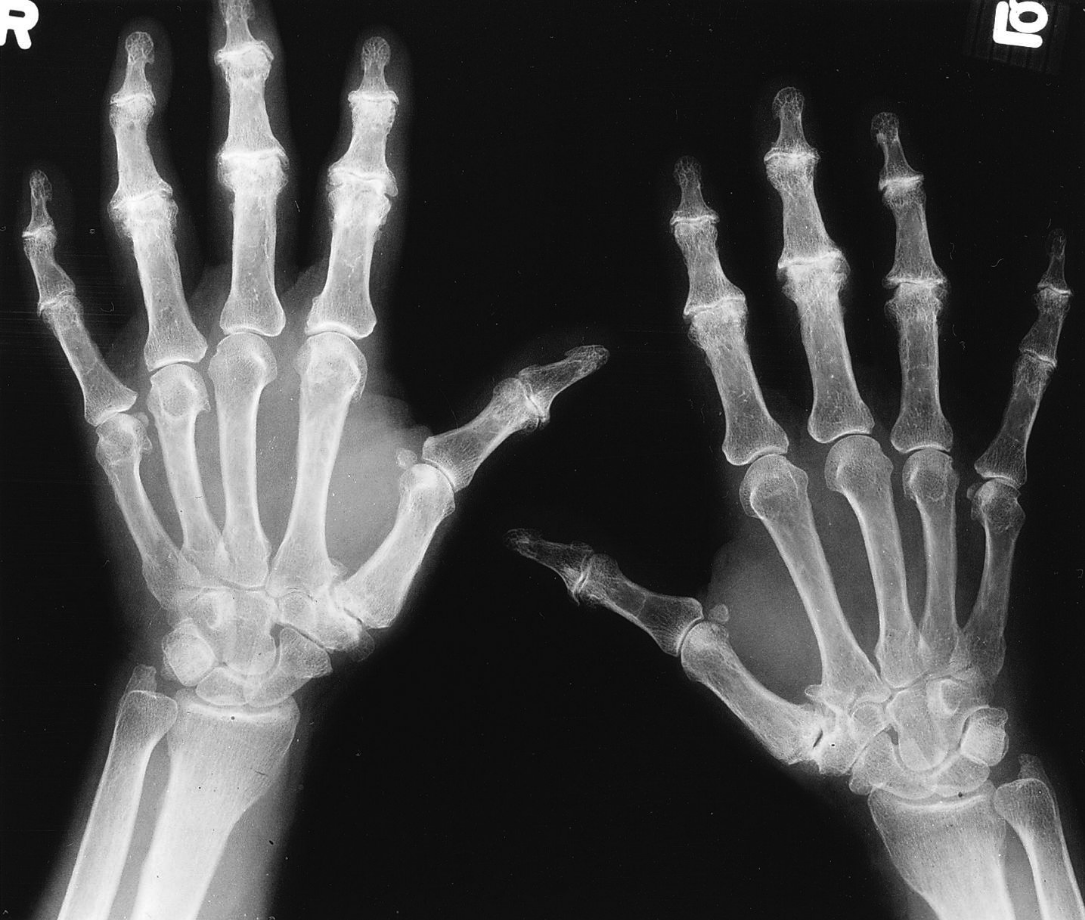
What is seen on imaging and what disease process is it a characteristic finding of?
“hook” osteophytes, particularly of the second and third MCP joints, are a characteristic finding in patients with hereditary hemochromatosis.
500
Though patients with HIT are thrombocytopenic, it is important to remember that they are actually in an overall prothrombotic state and thus require continued anticoagulation. List at least 3 anticoagulants that can be used as initial alternatives for heparin products in patients with HIT. Additionally, which common anticoagulant should not be used as an initial alternative and why?
 Factor Xa Inhibitors: Apixaban (Eliquis), Rivaroxaban (Xarelto), Fondaparinux (Arixtra)
Factor Xa Inhibitors: Apixaban (Eliquis), Rivaroxaban (Xarelto), Fondaparinux (Arixtra)
Direct Thrombin Inhibitors: Argatroban, Bivalirudin, Dabigatran
Warfarin should not be used as an initial alternative to heparin products as its antithrombotic efficacy takes 3-5 days to take effect, during which time patients are considered to be at increased risk of thrombosis due to decreased levels of Protein C.
500
TTP is associated with a high mortality rate and as such urgent therapy with Plasma Exchange, steroids, and rituximab can be life saving. Due to the urgent need for therapy, treatment is often started with a presumptive diagnosis of TTP as confirmatory tests, such as an ADAMTS13 activity level of < 10% or high ADAMTS13 inhibitor titers, may take too long to result. Which scoring system can help to guide clinical decision making when used in conjunction with appropriate history, physical exam, and lab evaluation?
The PLASMIC score can be used to judge the probability of TTP. A point is given for each of the following and a score of 0-4 indicates a low probability of TTP, a score of 5 indicates an intermediate probability of TTP, and a score of 6-7 indicates a high probability of TTP.
● Platelet count <30,000/microL
● Hemolysis (defined by reticulocyte count >2.5 percent, undetectable haptoglobin, or indirect bilirubin >2 mg/dL)
● No active cancer
● No solid organ or stem cell transplant
● MCV <90 fL
● INR <1.5
● Creatinine <2.0 mg/dL
*** the scoring system should be used in patients with thrombocytopenia and MAHA (schistocytes on peripheral smear) but should not be the sole determinant for initiation/discontinuation of presumptive therapy***
500
How is a diagnosis of Thrombotic thrombocytopenic purpura (TTP) made?
What is the treatment and when should it be initiated?
Testing for ADAMTS13 activity is important to confirm the diagnosis and establish the autoimmune cause. However, the turn-around time for this test result is long, and therapy should not be delayed while awaiting results.
The presence of schistocytes on the peripheral blood smear will confirm the diagnosis of a microangiopathic hemolytic anemia (MAHA) and a presumptive diagnosis of TTP and allows for early treatment.
Patients require emergent treatment with plasma exchange. Prompt diagnosis is critical because TTP is fatal in 90% of patients without therapy.
500
How does atypical HUS differ from classic HUS and what medication should be added to therapy if a severe case is suspected?
Atypical HUS with the same triad of thrombocytopenia, MAHA, and AKI as classic HUS but without the preceding diarrheal illness. It is thought to be caused by complement dysregulation caused by various triggers including drugs, SLE, or other infections such as HIV or streptococcus pneumoniae.
When atypical (or complement mediated) HUS is suspected, eculizumab, a monoclonal antibody that binds to complement C5 and prevents generation of the membrane attack unit C5b-9, should be added to the patient's therapy within 24-48 hours to limit irreversible renal injury.