Stroke
Neuromuscular
Movement Disorder
Epilepsy
Neuro-immunology
100
Full form of ABCD2 score
1. Age 2. Blood Pressure 3. Clinical Features 4. Duration of symptoms 5. Diabetes
Explanation
- The ABCD2 score should not be used as a substitute for clinical judgement or decision making.
- In populations with a low baseline stroke risk, a low ABCD2 score (0-2) correlates with a less than 1% risk of stroke within the next week. These patients may be appropriate for expedited outpatient evaluation.
- In centers where computed tomography and carotid ultrasound studies are performed in collaboration with a stroke service, adding the ABCD2 score to the evaluation appears unnecessary
100
Inheritance pattern of Duchenne and Becker Muscular Dystrophy
X linked Inheritance
100
Name two Synucleinopathies
1. Parkinson's Disease 2. Dementia with Lewy Body 3. Multiple System Atrophy
100
Patient has a triad of symptoms: (1)multiple generalized seizure types, including tonic, atonic, myoclonic, and atypical absence (2) an interictal EEG pattern of diffuse slow spike-wave complexes (3) cognitive dysfunction What is the diagnosis?
Lennox-Gastaut syndrome
Explanation
This syndrome is characterized by the presence multiple types of intractable seizures (in particular tonic seizures in sleep, but atonicand atypical absence seizures also occur), cognitive and behavioral impairments and diffuse slow spike-and-wave and paroxysms of fast activity on EEG.
NOTE Lennox Gastaut syndrome is considered an 'epileptic encephalopathy'. This term denotes the concept that the epileptic activity itself might directly contribute additional cognitive and behavioral impairments over those expected from the underlying etiology alone, and that suppression of epileptic activity might minimize this additional impairment.
Clinical context
This syndrome is characterized by onset of seizures from age 1 to 7 years (peak 3 to 5 years). Both sexes are affected. Antecedent, birth and neonatal history may be normal or there may be a history related to a structural brain abnormality (developmental or acquired). Neurological exam and head circumference may be normal or may reflect underlying structural brain abnormality, if present. Development and cognition prior to presentation is usually abnormal, but occasionally onset may occur in an otherwise normally developing child. Subsequent developmental stagnation or regression is typical after the onset of seizures.
Around 10-30% of cases of Lennox Gastaut syndrome evolve from earlier onset epilepsy syndromes, including West and Ohtahara syndromes. Occasionally there is a history of previous febrile seizures, focal seizures or generalized seizures.
Causes:
- Structural brain abnormalities (most common cause, 70% of cases)
- Genetic etiologies (de novo mutations) are expected to account for many of the currently unexplained cases
100
Two drugs used for Aggressive Multiple Sclerosis
1. Alemtuzumab 2. Mitoxantrone 2. Cladribine 4. Cyclophosphamide
200
HAMLET trial is related to
Malignant Middle cerebral artery infarction
Explanation
Malignant MCA stroke is indicated by:
- MCA territory stroke of >50% on CT
- Perfusion deficit of >66% on CT
- Infarct volume >82 mL within 6 hours of onset (on MRI)
- Infarct volume of >145mL within 14 hours of onset (on MRI)
- There are 3 important trials that have studied decompressive hemicraniectomy for malignant MCA strokes in patients <60 years of age
DESTINY trial (2007)
- Prospective, MC RCT from Germany
- n=32 (projected sample size calculated to be n-188)
- steering committee terminated the trial early as a statistically significant mortality reduction was found at this stage in comibnation with the results of the other European decompressive craniectomy trials
- Outcome: 88% vs 47% survival in favour of decompressive craniectomy
DECIMAL trial (2007)
- Prospective, MC RCT from France
- n=38
- data safety monitoring committee terminated the trial because of slow recruitment
- Outcome: ARR 52.8% in mortality favouring the decompressive craniectomy group (75% vs 22% survival)
HAMLET trial (2009)
- Prospective, MC RCT from the Netherlands
- n=64
- Outcome: ARR 38% in mortality favouring the decompressive craniectomy group
Pooled analysis of DESTINY, DECIMAL and HAMLET (Vahedi et al, 2007)
- n=93
- Patients aged <60y with supratentorial infarctions treated with decompressive craniectomy, usually within 48 hours of stroke onset
- With hemicraniectomy compared with medical management:
- Reduced mortality (22% versus 71% – pooled analysis; NNT=2)
- No individual study showed an improvement in the percentage of survivors with good outcomes (mRS score, 0–3)
- Only shown in a pooled analysis (43% versus 21%).
- Only 14% of surgical survivors could look after their own affairs without assistance (mRS score, 2)
- no difference in outcome whether dominant or non-dominant hemispheres are involved
Subsequently, the DESTINY II Trial (2014) studied patients aged >60 years:
- n = 112 patients >60 years of age (median age was 70)
- Primary outcome measure was survival without severe disability
- 38% in the hemicraniectomy group vs 18% in the control group
- Secondary outcomes:
- Overall mortality was lower in the surgery group (33% vs 70%)
- Almost none of the survivors has an outcome as good as an mRS score of 3; almost all post-operative survivors were severely disabled
AN APPROACH
Decompressive hemicraniectomy
- can be considered in patients <60 years of age, within 48 hours of stroke onset, although outcomes are still likely to be poor
- should not be performed in malignant MCA stroke patients aged >60 years as survivors will be severely disabled
200
Which type of muscle fiber atrophy is characteristic of steroid myopathy?
Type 2 muscle fiber
Explanation
Steroid myopathy may be more frequent with the use of fluorinated steroids, such as dexamethasone or triamcinolone, than with nonfluorinated ones, such as prednisone or hydrocortisone.
200
The rocket sign occurs in patients with which Parkinsonian disorder ?
Progressive Supranuclear Palsy
200
Drug of choice for infantile spasms in a patient with Tuberous Sclerosis
Vigabatrin
Explanation
In 1841, Dr. William James West wrote a letter to Lancet describing new infantile convulsions in his 4-month-old son. His apt description of the events, starting with a subtle head drop and progressing to clusters of myoclonic spasms occurring many times daily, accompanied by the regression of development of his previously normal child, remains the hallmark of this condition today. His letter also embodies the distress of a father and medical provider caused by this rare, difficult-to-treat, and frequently, neurologically devastating condition
200
Oral Multiple Sclerosis drug with absolute contraindication during pregnancy
Teriflunomide
300
Final conclusion of CHANCE trial
Patients with TIA who can initiate antiplatelets within 24 hours after symptom onset combination of clopidogrel and aspirin for the first 21 days is superior to aspirin alone for reducing stroke in first 90 days
300
Which antibodies are seen in as many as two-thirds of patients with Inclusion Body Myositis?
Cytosolic 5'-nucleotidase 1A
Explanation
It is rare for healthy adults to have a positive NT5C1A antibody test. The test is sometimes positive in some other autoimmune disease, such as dermatomyositis, systemic lupus erythematosus, and Sjogren’s syndrome (~10-25% positives). However, these diseases can usually be distinguished from IBM based on the patient’s history and examination.
Polymyositis is often confused with inclusion body myositis. The antibody test can help separate the two conditions, as the test is rarely positive (<5%) in polymyositis.
300
Molar tooth sign on MRI is seen in
Joubert Syndrome
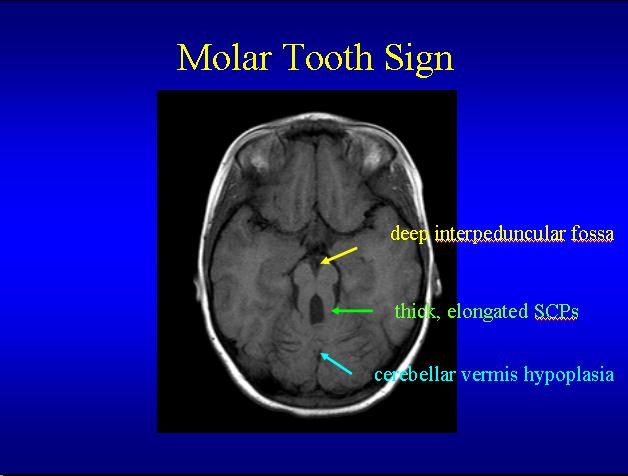
300
Which syndrome is characterized by the onset in early childhood of focal autonomic seizures that are often prolonged
Panayiotopoulos
Panayiotopoulos syndrome is characterized by onset of seizures between 1 and 14 years of age (majority between 3 and 6 years). Seizures are infrequent in most patients, with 25% having a single seizure (which may be autonomic status epilepticus) and 50% having six seizures or less. Frequent seizures can occur in some patients. Seizures usually resolve by age 11-13 years. Both sexes are affected equally. Antecedent and birth history is normal. Head size and neurological examination are usually normal. Development and cognition are normal. However, during active seizure periods, subtle neuropsychological deficits in language and executive functioning have been reported. A history of febrile seizures is seen in 5-17% of patients.
300
Mechanism of action of Fingolimod
Interacts with Sphingosone-1-phosphate receptor
400
Results of CLOSE trial published in 2017
Among patients with cryptogenic stroke with PFO rate of stroke occurrence in lower among those assigned to PFO closure combined with anti platelet therapy compared to those with anti platelet alone
The following are key points to remember from this review of cryptogenic stroke and patent foramen ovale (PFO):
- PFO is associated with cryptogenic stroke (stroke of unclear etiology). PFO is present in 20-25% of the adult population, but in 40% of adults with cryptogenic stroke. The current article serves to summarize the history and present state of PFO closure for secondary stroke prevention.
- Despite the association between PFO and cryptogenic stroke, three early randomized clinical trials (CLOSURE I, PC trial, and RESPECT short-term) did not show a clear benefit of PFO closure for secondary stroke prevention. These results led to a generally decreased interest in PFO closure for stroke prevention.
- In March 2016, a meta-analysis of patient-level data from CLOSURE I, PC, and RESPECT was published. This meta-analysis found that PFO closure was superior to medical therapy for the prevention of recurrent ischemic stroke (adjusted hazard ratio [aHR], 0.58; 95% confidence interval [CI], 0.34-0.99). When the analysis was restricted to the trials in which only the Amplatzer PFO occluder device was used (PC and RESPECT), the benefit appeared even greater (aHR, 0.41; 95% CI, 0.20-0.88).
- In October 2016, the Food and Drug Administration (FDA) approved the Amplatzer PFO occluder device for patients 18-60 years old with PFO and cryptogenic stroke.
- In the last year, two new randomized controlled trials of PFO closure versus medical therapy were published: CLOSE and REDUCE. These trials used stricter enrollment criteria than the three previous trials.
- In CLOSE, eligible patients were 16-60 years old, had had a cryptogenic stroke with corroborating imaging findings within the prior 6 months, had a PFO, and had a large interatrial shunt or atrial septal aneurysm. Patients could not have small vessel disease or ≥30% stenosis of an artery supplying the brain. After a mean follow-up of 5.3 years, subjects who underwent PFO closure had a lower risk of recurrent stroke than those maintained on antiplatelet therapy (0% vs. 6%; HR, 0.03; 95% CI, 0-0.26).
- The data from CLOSE suggest that, for every 20 closed patients, one stroke is avoided at 5 years. However, the avoided stroke may not be a disabling stroke. No patients (of 238) in the closure group had a disabling stroke; 1 patient (of 235) in the antiplatelet group had a disabling stroke.
- In REDUCE, eligible patients were 18-59 years, had had a recent cryptogenic stroke within the prior 6 months, had symptoms lasting ≥24 hours or positive imaging, and had a PFO. Patients could not have a stenosis of ≥50% of a major vessel, a lacunar stroke, or uncontrolled stroke risk factors. After a median follow-up of 3.2 years, subjects who underwent PFO closure had a lower risk of recurrent stroke than those maintained on antiplatelet therapy (1.4% vs. 5.4%; HR, 0.23; 95% CI, 0.09-0.62).
- The data from REDUCE suggest that, for every 28 closed patients, one stroke is avoided at 2 years.
- In both REDUCE and CLOSE, PFO closure was associated with a higher risk of atrial fibrillation, which was believed to be primarily due to the closure procedure itself (i.e., self-limited).
- CLOSE and REDUCE were likely positive because of stricter enrollment criteria, leading to the inclusion of subjects whose presenting stroke was secondary to PFO rather than another etiology (such as large artery atherosclerosis, small vessel disease, or atrial fibrillation).
- In summary, PFO closure is of moderate benefit compared to antiplatelet therapy alone in the prevention of recurrent ischemic stroke in adults up to 60 years of age. It remains unknown how PFO closure compares to systemic anticoagulation (e.g., with novel oral anticoagulants) for the prevention of recurrent ischemic stroke.
400
Which muscular dystrophy is triad of (1) contractures of the elbow flexors, ankle plantar flexors, and spine early in disease (2) childhood onset of humeroperoneal weakness (3) cardiac disease with arrhythmias, conduction block, and cardiomyopathy
Emery-Dreifuss muscular dystrophy
Emery-Dreifuss muscular dystrophy (EDMD) should be suspected in individuals with the following triad:
Early contractures of the elbow flexors, Achilles tendons (heels), and neck extensors resulting in limitation of neck flexion, followed by limitation of extension of the entire spine
Slowly progressive wasting and weakness typically of the humero-peroneal/scapulo-peroneal muscles in the early stages
Cardiac disease with conduction defects and arrhythmias
Atrial fibrillation, flutter and standstill, supraventricular and ventricular arrhythmias, and atrio-ventricular and bundle-branch blocks may be identified on resting electrocardiography (ECG) or by 24-hour ambulatory ECG.
Dilated or hypertrophic cardiomyopathy may be detected by the performance of echocardiographic evaluation.
400
76-year-old man showing increased signal intensity in the middle cerebellar peduncles. The patient presented to a movement disorders clinic with progressive gait ataxia since the age of 68, with later development of memory problems, occasional confusion, and tremor in his left hand during walking. His family history revealed developmental disability in his grandson through his daughter and premature ovarian failure in one of his granddaughters
Fragile X tremor-ataxia syndrome
400
4 year old child comes with episode of a brief generalized myoclonic jerk affecting proximal muscles, and is followed by abrupt fall. What is the diagnosis?
Myoclonic- Atonic Seizure. Doose Syndrome
This syndrome is characterized by seizures that have onset between 6 months and 6 years of age (peak 2 to 4 years). In two thirds of children febrile seizures and generalized tonic-clonic seizures precede the onset of myoclonic-atonic and atonic seizures. Both sexes are affected, with a male predominance (ratio 2:1). Antecedent and birth history is unremarkable. Neurological examination and head size are normal. Development and cognition is typically normal, however impairments may develop at or after seizure onset.
500
A 21-year-old man presented to the emergency department reporting bilateral blurry vision. The visual symptoms began abruptly 2 days earlier and were preceded by a thunderclap headache the week before. A right homonymous hemianopia was found on confrontational visual field testing. He was not able to read simple or complex sentences. . Laboratory testing was remarkable for a serum creatinine of 3.2 mg/dL, and ECG demonstrated a right bundle branch block. Brain MRI revealed multifocal areas of acute infarction predominantly involving the vertebrobasilar circulation. Patient’s sister had developed progressive visual deterioration and painful acroparesthesias during adolescence. What is the diagnostic blood test for this patient?
Alpha Galactosidase (Fabry's Disease)
Explanation
Clinical research programs of enzyme replacement therapy (ERT) for Fabry disease led to commercial availability of agalsidase alpha (Replagal®, Shire Human Genetic Therapies, Inc.) and agalsidase beta (Fabrazyme®, Genzyme Corp.) in most European countries in 2001, and for agalsidase beta in the US in 2003.
Adult males (?16 y) At time of diagnosis of Fabry disease
Pediatric males At time of development of significant symptomsa or If asymptomatic, consider at 10–13 yr
Females (all ages) Monitor; institute if significant symptomsa or evidence of progression of organ involvement
500
Andersen-Tawil syndrome is characterized by the clinical triad
(1) episodic flaccid muscle weakness (2) ventricular arrhythmias and prolonged QT interval (3) dysmorphic features
Explanation
Andersen-Tawil syndrome is a disorder that causes episodes of muscle weakness (periodic paralysis), changes in heart rhythm (arrhythmia), and developmental abnormalities. Periodic paralysis begins early in life, and episodes last from hours to days. These episodes may occur after exercise or long periods of rest, but they often have no obvious trigger. Muscle strength usually returns to normal between episodes. However, mild muscle weakness may eventually become permanent.
500
A 79-year-old man had a 7-year course of markedly asymmetric parkinsonism and progressive aphasia. At onset, his family noticed frequent jerks of his left arm and leg. His balance worsened, and he tended to lean to the left and stall with gait freezing. He had trouble figuring out how to sit down on a toilet seat and needed assistance with dressing, grooming, and eating. His left hand would sometimes wander and go into his pocket or grab his shirt without his knowledge. He had no response to levodopa. What is diagnosis?
Cortico Basal Syndrome
500
Patient presents with progressive myoclonic epilepsy, Ataxia, mild cognitive deterioration, generalized tonic-clonic seizures. Genetic testing reveals EPM1 abnormality. What is the diagnosis?
Unverricht Lundborg disease
Explanation
Progressive myoclonus epilepsy should considered in a patient with myoclonic seizures, with or without generalized tonic-clonic seizures in the following settings:
- Progressive cognitive decline
- Myoclonus resulting in progressive motor impairment
- Cerebellar signs
- Background slowing on EEG (particularly if increasing over time)
- Myoclonus that is refractory to trials of appropriate anti-seizure medication
Causes
Many metabolic and genetic causes of progressive myoclonus epilepsy exist, and specific etiologies often present at certain ages. However, despite extensive evaluation, the cause of progressive myoclonus epilepsy may remain unknown in some patients. Causes that are recognized include:
Neurodegenerative conditions: Unverricht-Lundborg disease and Lafora disease.
Less commonly: Dentatorubo-pallidoluysian atrophy, juvenile Huntingtons, action-myoclonus-renal failure syndrome, juvenile neuroaxonal dystrophy, pantothenate-kinase associated neurodegeneration, neuroserpin inclusion body disease, leukoencephalopathy with vanishing white matter, early-onset Alzheimers and GOSR2 mutations.
Metabolic etiologies: mitochondrial disorders (MERRF), neuronal ceroid lipofuscinosis, sialidosis.
Less commonly: mitochondrial disorders (POLG1, MELAS, others), GM2 gangliosidoses, tetrahydrobiopterin deficiency, non-infantile neuronopathic Gaucher's disease and Niemann Pick type C.
- Immune etiologies: celiac disease (uncommon)