Mission Bay
The Dogpatch
Bernal Heights
Outer Sunset
Hayes Valley
100
One mutant allele at the LDL receptor locus (leading to familial hypercholesterolemia) encodes an elongated protein that is approximately 50,000 Da larger than the normal 120,000-Da receptor.
Indicate at least three mechanisms that could account for this abnormality.
Approximately how many extra nucleotides would need to be translated to add 50,000 Da to the protein?
Three types of mutations could explain a mutant protein that is 50 kD larger than the normal polypeptide:
1. A mutation in the normal stop codon that allows translation to continue.
2. A splice mutation that results in the inclusion of intron sequences in the coding region. The intron sequences would have to be free of stop codons for sufficient length to allow the extra 50 kD of translation.
3. An insertion, with an open reading frame, into the coding sequence.
For any of these, approximately 500 extra residues would be added to the protein if the average molecular weight of an amino acid is approximately 100.
Five hundred amino acids would be encoded by 1500 nucleotides.
100
Mrs. Soft has a history of multiple fractures, mild short stature and hearing loss. She brings her two-month-old son, Carl, to you for evaluation. Carl has no history of fractures. He has a normal physical exam. Which of the following evaluations is the most accurate way to determine if Carl has the same condition as his mother?
A) Determine if Carl has a mutation in his COL1A1 or COL1A2 genes
B) Determine if Carl has an abnormal ABD or OAE test
C) Determine if Carl's skull radiographs demonstrate Wormian bones
D) Determine if Mrs. Soft and Carl share a COL1A1/COL1A2 mutation
E) Determine if radiographs of Carl's extremity bones show fractures
D) Determine if Mrs. Soft and Carl share a COL1A1/COL1A2 mutation
Mrs. Soft most likely has Osteogenesis Imperfecta (OI) type 1 which is caused by missense mutations, small insertions or deletions, or exon-skipping mutations of COL1A1 or COL1A2. You should test Mrs. Soft to confirm that she has a detectable COL1A1 or COL1A2 OI causing mutation. You can then test her son for her mutation. If he is positive, then he is at great risk to develop findings of OI. If he is negative for the mutation, then he is at population risk to develop OI. a) His not having an OI causing COL1A1 or COL1A2 mutation could result from Mrs. Soft not having OI Type 1, he has her mutation but it is not detectable, or he does not have her mutation. Thus if he has a negative result you are in doubt. b, c and e) These options are all looking for pleiotropic effects of OI that can have reduced penetrance and thus could be negative at two months of age.
100
You provide a clinical description of a patient to the molecular cytogeneticist at your facility including: a prominent nasal root, bulbous nasal tip, hypocalcemia, immunodeficiency, and conotruncal heart abnormality. Which of the following laboratory techniques is best used to confirm the suspected diagnosis in this patient?
A) Next generation sequencing
B) Routine cytogenetic testing
C) Southern Blot
D) Flourescence-in-situ hybridization or DNA microarray
E) Sanger sequencing of the TBX1 gene
D) Flourescence-in-situ hybridization or DNA microarray
Explanation: The clinical description is specific and indicative of a targeted cytogenetic abnormality, which can be identified by FISH analysis. Methodologies such as next generation, routine cytogenetic testing are useful when a specific condition is not indicated from the clinical presentation. Southern blot analysis is labor intensive method to detect the abnormality whereas sequencing will not detect the common mutation in the indicated abnormality.
100
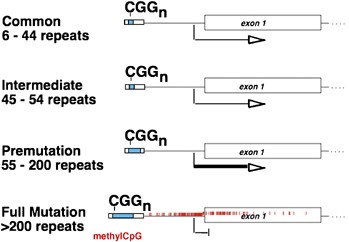
A 60-year-old man presents to neurology clinic with a history of late-onset, progressive cerebellar ataxia and intention tremor. A family history reveals his daughter has a son with fragile X syndrome. Which of the following alterations in the FMR1 gene is most likely to be found in this patient?
A) Point mutation
B) 30 CGG repeats
C) 80 CGG repeats
D) 250 CGG repeats
E) Deletion of the FMR1 gene
C) 80 CGG repeats
Fragile X tremor/ataxia syndrome occurs in males with FMR1 premutation alleles.
100
You evaluate a 3-day-old boy with an abnormal Newborn Screening (NBS) result. He has a 2-day history of poor feeding, emesis and diarrhea. On physical examination he is somewhat icteric and has hepatomegaly. Which of the following disorders is most likely to be implicated by the abnormal NBS result?
A) Cystic fibrosis
B) Galactosemia
C) Homocystinuria
D) Pendred syndrome
E) Phenylketonuria
B) Galactosemia
This neonate has poor feeding, emesis, diarrhea, icterus and hepatomegaly. The early onset and all of these signs and symptoms are seen in galactosemia. A. Cystic Fibrosis can present with diarrhea but not usually with poor feeding, emesis or hepatomegaly. C. Homocystinuria is usually asymptomatic in neonates. D. Pendred Syndrome can present with hearing loss but not usually with poor feeding, emesis or hepatomegaly. E. Phenylketonuria is usually asymptomatic in neonates.
200
Are autosomal dominant PSCK9 gain-of-function mutations that cause familial hypercholesterolemia deficiency phenocopies, or genocopies, of familial hypercholesterolemia due to autosomal dominant mutations in the LDL receptor gene?
Explain your answer.
Autosomal dominant PCSK9 gain-of-function mutations that cause familial hypercholesterolemia are genocopies of autosomal dominant loss-of-function mutations in the LDL receptor gene (LDLR), because a genocopy is a genotype that determines a phenotype closely similar to that determined by a different genotype (for comparison, see Glossary for the definition of phenocopy).
200
DMD has a high mutation rate but shows no ethnic variation in frequency. Use your knowledge of the gene and the genetics of DMD to suggest why this disorder is equally common in all populations.
For DMD, as a classic X-linked recessive disease that is lethal in males, one third of cases are predicted to be new mutations. The large size of the gene is likely to account for the high mutation rate at this locus (i.e., it is a large target for mutation). The ethnic origin of the patient will have no effect on either of these phenomena.
200
You are following a 30-year-old pregnant woman with von Hippel-Lindau syndrome. Which of the following findings is the most important to monitor her for during the pregnancy?
A) Endolymphatic sac tumor
B) Pheochromocytoma
C) Renal Cancer
D) Retinal hemangioblastoma
E) Vestibular schwanomma
B) Pheochromocytoma
Explanation: Women with VHL should be followed during pregnancy for development of pheochromocytoma. The other matters are not specifically critical during pregnancy, and vestibular schwannoma is not a feature of VHL.
200
A male is found to have 400 CGG repeats. Which of the following disorders is the most likely diagnosis?
A) Fragile X
B) Friedreich ataxia
C) Fragile X-associated tremor/ataxia syndrome (FXTAS)
D) Huntington disease
E) Myotonic dystrophy
The only CGG expansion disorders in this list is Fragile X and FXTAS. A large expansion of this size would lead to FRAXA, while a premutation would cause FXTAS. The size of the CGG repeat is consistent with Fragile X symptoms, not FXTAS. Myotonic is CTG, FXTAS

200
An asymptomatic infant who is found to have hyperphenylalaninemia on newborn screening should be tested for which of the following associated problems?
A) biopterin synthesis defect
B) catechol-o-methyltransferase
C) liver disease
D) microdeletion deficiency of chromosome 12
E) porphyria cutanea tarda
A) biopterin synthesis defect
Tetrahydrobiopterin is a cofactor for phenylalanine hydroxylase (along with oxygen) that converts phe to tyr ~2% of infants with inherited hnyperphenylalaninemia have a defect in the synthesis or recycling of the biopterin cofactor. All infants with confirmed hyperphe should be tested for a biopterin defect by blood and urine pterin screening.

300
Johnny, 2 years of age, is failing to thrive. Investigations show that although he has clinical findings of CF, his sweat chloride concentration is normal. The sweat chloride concentration is normal in less than 2% of patients with CF. His pediatrician and parents want to know if DNA analysis can determine whether he indeed has CF.
a. Would DNA analysis be useful in this case? Briefly outline the steps involved in obtaining a DNA diagnosis for CF.
b. If he has CF, what is the probability that he is homozygous for the ΔF508 mutation? (Assume that 85% of CF mutations could be detected at the time you are consulted and that his parents are from northern Europe, where the ΔF508 allele has a frequency of 0.70.)
c. If he does not have the ΔF508 mutation, does this disprove the diagnosis? Explain.
If Johnny has CF, the chances are approximately 0.85 × 0.85, or 70%, that he has a previously described mutation that could be readily identified by DNA analysis. His parents are from northern Europe; therefore the probability that he is homozygous for the ΔF508 mutation is 0.7 × 0.7, or 50%, because approximately 70% of CF carriers in northern Europe have this mutation. If he does not have the ΔF508 mutation, he could certainly still have CF, because approximately 30% of the alleles (in the northern European population, at least) are not ΔF508. Steps to DNA diagnosis for CF include the following: (1) look directly for the ΔF508 mutation; if it is not present, (2) look for other mutations common in the specific population; (3) then look directly for other mutations based on probabilities suggested by the haplotype data; (4) if all efforts to identify a mutation fail (or if time does not allow), perform linkage analysis with polymorphic DNA markers closely linked to CF.
300
Which of the following NBS acylcarnitine profiles is most suggestive of MCAD deficiency?
A) Increased C3 and C4
B) Increased C5-OH
C) Increased C6, C8 and C8/ C10 ratio
D) Increased C14-OH, C16-OH, C18-OH and C18:1-OH
E) Increased C14:1 and C14:1/ C12:1 ratio
C) Increased C6, C8 and C8/ C10 ratio
Increased C6, C8 and C8/ C10 ratio is the profile for MCAD and these are derived from medium chain fatty acids. Increased C14-OH, C16-OH, C18-OH and C18:1-OH is the profile for LCHAD Deficiency. Increased C14:1 and C14:1/ C12:1 ratio is the profile for VLCAD deficiency. Increased C3 and C4 These are seen in Propionic academia and SCAD deficiency, respectively.
300
A 50 year old man presents with pain and is found to have multiple schwannomas. A brain MRI shows no evidence for vestibular schwannoma. Which pair of genes would be appropriate to test to determine whether he might have schwannomatosis?
A) NF1 and NF2
B) NF2 and TSC2
C) NF1 and TSC2
D) TSC2 and LZTR1
E) SMARCB1 and LZTR1
E) SMARCB1 and LZTR1
Explanation: SMARCB1 and LZTR1 are both associated with schwannomatosis. Schwannomas can be found in NF2 patients, but usually in association with vestibular schwannomas and other features. TSC2 is associated with tuberous sclerosis complex, not schwannomatosis.
300
The CTG repeat expansion responsible for myotonic dystrophy affects which of the following molecular processes for the DMPK gene or gene product?
A) Level of transcription
B) Protein degradation in the proteosome
C) RNA binding protein
D) RNA editing
RNA binding protein
300
A 6-month-old infant presents with FTT, enlarged liver, hypotonia, and developmental delay. You diagnose Carbohydrate Deficient Glycoprotein syndrome Type Ib based on transferrin glycosylation and gene sequencing. Which of the following interventions is the most appropriate treatment for this disorder?
A) Enzyme replacement therapy
B) Oral mannose supplementation
C) Supportive care only
D) Bone marrow transplant
E) Low carbohydrate elemental formula
B) Oral mannose supplementation
CDG Type Ib is the only type currently known with a specific treatment. The defect is mannose-6-phosphate isomerase.
400
James is the only person in his kindred affected by DMD. He has one unaffected brother, Joe. DNA analysis shows that James has a deletion in the DMD gene and that Joe has received the same maternal X chromosome, but one without a deletion. What genetic counseling would you give the parents regarding the recurrence risk for DMD in a future pregnancy?
James may have a new mutation on the X chromosome because Joe inherited the same X chromosome from his mother, and the deletion was present in neither Joe nor his mother. If this is the case, there is no risk for recurrence. Alternatively, the mother may be a mosaic, and the mosaicism includes her germline. In this case, there is a definite risk that the mutant X could be inherited by another son or passed to a carrier daughter. Approximately 5% to 15% of cases of this type appear to be due to maternal germline mosaicism. Thus the risk is half of this figure for her male offspring because the chance that a son will inherit the mutant X is × 5% to 15% = 2.5% to 7.5%.
400
Which of the following disorders is associated with duplication of gene on chromosome 17?
A) Charcot-Marie-Tooth disease
B) Hereditary liability to pressure palsies
C) Miller-Dieker syndrome
D) NF1
E) Spinal muscular atrophy
A) Charcot-Marie-Tooth disease
Chariot-Marie Tooth disease is associated with PMP22 duplication; hereditary neuropathy with liability to pressure palsies is associated with deletion of this gene.
400
A clinical laboratory has received funding from their institution to implement next generation sequencing in their laboratory. The laboratory is tasked to evaluate the instruments and sequencing chemistries on the market and provide a justification for selection of a particular instrument and chemistry. Amongst the parameters being evaluated the laboratory is looking at Phred scores. A Q40 Phred Score reflects which of the following likelihoods that the base call is inaccurate.
A) 1:40
B) 1:4,000
C) 1:100
D) 1:1,000
E) 1:10,000
E) 1:10,000
Explanation: Phred scores are indicative of the quality of the sequence data and the higher the Phred score lower the error rate in the sequence data. A Phred score of Q40 is considered the highest and best Phred indicative of an error rate of 1:10,000 in the sequence data.
400
An individual is found to have a 33 and a 300 GAA repeat. Which of the following results is the most likely interpretation?
A) Affected with Huntington disease
B) Affected with Friedreich ataxia
C) Affected with myotonic dystrophy
D) Carrier of Friedreich ataxia
E) Female carrier of Fragile X with symptoms
D) Carrier of Friedreich ataxia
Only 4% of FRDA patients will have an expansion and a point mutation. Most FRDA carriers will have an expansion and a normal allele. If this were a symptomatic individual, one would recommend the point mutation analysis. For diagnostic testing, finding one expansion confirms that the individual is at least a carrier but does not confirm the diagnosis, although it is strongly suggestive.
400
A newborn infant is diagnosed with Medium Chain AcylCoA Dehydrogenase Deficiency (MCADD) based on newborn screening (NBS) results. While awaiting the molecular analysis to confirm the NBS result, which of the following complications should you warn the parents about during their initial visit to your medical genetics clinic?
A) Hyperammonemia
B) Hypoglycemia
C) Ketosis
D) Neutropenia
E) Thrombocytopenia
B) Hypoglycemia
Explanation: Children with MCADD who are fasted or have poor po intake are prone to develop hypoketotic hypoglycemia. Before NBS for MCADD the first crisis was fatal in 25% of affected children. The other answers are not typical for MCADD.
500
A 3.5-year-old girl, T.N., has been noted to have increasing difficulty standing up after sitting on the floor. Her serum level of creatine kinase is grossly elevated. Although a female, the presumptive clinical diagnosis is Duchenne muscular dystrophy. Females with DMD are rare. Identify three mechanisms of mutation that could account for the occurrence of DMD in a female.
A DMD female like T.N. might have the disease because she carries a DMD gene mutation on the X chromosome inherited from her mother. T.N. could show clinical symptoms if her paternal X (carrying a normal allele at this locus) was subject to nonrandom inactivation in most or all cells. An alternative explanation would be that she has Turner syndrome and that her only X chromosome (inherited from her mother) carries a DMD gene mutation. A third explanation would be that she has a balanced X;autosome translocation that disrupts the DMD gene on the translocated X chromosome. Although her normal X chromosome carries a normal allele at the DMD locus, balanced X;autosome translocations show nonrandom inactivation of the structurally normal X due to secondary cell selection.
500
Which of the following genes includes a polymorphism that is associated with risk of Alzheimer disease?
A) amyloid precursor protein
B) apoB-48
C) apoE
D) presenilin
E) prion protein
C) apoE
500
A DNA analysis using a dinucleotide repeat within a gene for an autosomal dominant disorder is presented below. The father and deceased grandfather are affected. . What is the probability that the pregnancy (P) is affected?
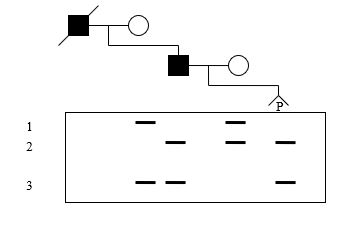
A) 1.0
B) 0.75
C) 0.5
D) 0.25
E) 0.0
E) 0.0
The phase in the father can be deduced to be allele 2 with the disease allele since both had to come from the grandfather. The fetus receives allele 3 from the father and allele 2 from the mother and is unaffected.
500
Mary's sister's 2-year-old son was recently diagnosed with Duchenne muscular dystrophy (DMD). He was tested by microarray analysis and found to have a deletion of exons 44-52. Mary is 6 weeks pregnant and wants to know her DMD carrier status. Her blood sample is sent to a different lab that uses MLPA for dystrophin carrier testing. The lab does not find the exon 44-52 deletion but instead finds an exon 3 deletion. Which of the following explanations most likely accounts for these results?
A) Mary's DNA has a variant at the probe binding site causing a false positive result.
B) Mary's mother is a germline mosaic for two different dystrophin mutations.
C) One of the labs had a sample mix up, misidentifying the sample.
D) The exon 3 deletion represents a new mutation that occurred in Mary.
E) The microarray analysis produced false positive results due to incomplete hybridization.
A) Mary's DNA has a variant at the probe binding site causing a false positive result.
The greatest technical concern about the MLPA is regarding SNPs at the probe site that would interfere with binding of the probe and ligation, which produce false positive results. Generally, this would result in only a single exon being apparently deleted. For this reason, single exon deletion results should be confirmed using another technique. While some of the distractors could also happen, by far, the most likely explanation is as described above.
500
As you evaluate a 4-day-old boy with a blood sugar of 37, an anion gap of 21, 4+ urinary ketones and an ammonia level of 197, the State Newborn Screening (NBS) Lab contacts you to report an emergency NBS result. The emergency result is most likely to reveal an increase in which of the following levels?
A) Biotinidase enzyme activity
B) C3 acylcarnitine
C) C6, C8 and C8/C10
D) GALT enzyme activity
E) Succinylacetone
B) C3 acylcarnitine
This neonate has hypoglycemia, ketoacidosis and hyperammonemia. These are all consistent with an organic acidemia. C3 acylcarnitine is elevated in methylmalonic acidemia and propionic acidemia. A. Deficiencies rather than increases in Biotinidase enzyme activity cause disease. C. Increases in these analytes are seen in MCAD deficiency which is usually not associated with ketoacidosis. D. Deficiencies rather than increases in GALT enzyme activity cause galactosemia. E. Increases in Succinylacetone are seen in tyrosinemia type 1 which usually doesn't present this early.