Clinical Application
Pathophysiology
Surprise!
Labs/Histo/
Management and Diagnostic
100
List at least 4 common characteristic features seen in patients with Type 1 (Von Gierke disease)
- Renomegaly and hepatomegaly (with hepatic adenomas)
- Severe fasting hypoglycemia, mild ketosis
- Severe hyperlipidemia (especially triglycerides) → doll-like facies
- Hyperuricemia (↑ risk of gout)
- Lactic acidosis
- Anemia
- Failure to thrive
- Dysfunctional glycogenolysis and gluconeogenesis
100
Name 3 organs most commonly affected by GSD
Liver, heart, and muscle are the most common sites of glycogen storage and are, therefore, predominantly affected.
100
What type of bond is seen in glycogen branches, what type of bond is seen in glycogen linear linkages?
Branches have α-(1,6) bonds; linear linkages have α-(1,4) bonds.
100
Would we see an increase or decrease in blood lactate levels, uric acid levels, and triglyceride levels in patients with Type 1 (Von Gierke Disease)?
Increase in all of them.
100
What is a biomarker used to detect liver GSD?
↑ Serum biotinidase serves as diagnostic biomarkers in type I and type III
200
What are some defects of muscle glycogenolysis and muscle glycolysis seen in patients with GSD
- Easy fatiguability, exercise intolerance
- Cramps
- Rhabdomyolysis → myoglobinuria (burgundy-colored urine)
200
Describe the pathophysiology of GSD (big picture sequence of events)
Defective enzymes responsible for glycolysis or glycogenolysis → impaired glycogen metabolization → ↑ storage of either normal or abnormal glycogen in various organs, leading to different clinical presentations
200
Oh did you think there would be a question here....? some points perhaps?
should've never picked this question...

200
What serum levels are seen in patients with Hers disease? what enzyme is deficient in this type?
- Liver phosphorylase is deficient in this type of GSD. Liver glycogen phosphorylase catalyzes the cleavage of 1,4 glucosidic bonds to release glucose 1-phosphate from glycogen
- Elevated concentrations of serum transaminase
- Hyperlipidemia
- Elevated Uric Acid levels seen in some patients
200
What initial and confirmatory tests can be done to detect GSD?
- Muscle and/or liver biopsy (depending on the enzyme deficiency): glycogen storage appears as PAS-positive granules .
- Enzyme assays in RBCs, leukocytes, liver tissue, muscle tissue, or fibroblasts (depending on the enzyme deficiency)
- DNA testing for the gene defects
- Electroneuromyography: to identify proximal myopathy
- EKG and/or echocardiography: to identify cardiac hypertrophy and conduction blocks
300
List some of the newborn screening tests mandated by Texas law
Every baby born in Texas gets two newborn screening blood tests that check for 28 specific metabolic, endocrine, hematologic, and genetic disorders at 24 to 48 hours after birth regardless of feeding status and/or before any transfusions. Every infant must receive another screening between 1 and 2 weeks of age, as some cases may only be detected in the second screen. These include
- Amino Acid Disorders
- Fatty Acid Oxidation Disorders
- Organic Acid Disorders
- Endocrine Disorders
- Hemoglobin Disorders
- Other Disorders
The first test is done 24 to 48 hours after birth. The second one is done at the baby’s checkup at one to two weeks of age. Finding and treating these disorders early can prevent serious complications such as growth and developmental delays, deafness, blindness, intellectual disabilities, seizures, and sudden or early death.
The Newborn Screening Program began screening for SMA on June 1, 2021.
300
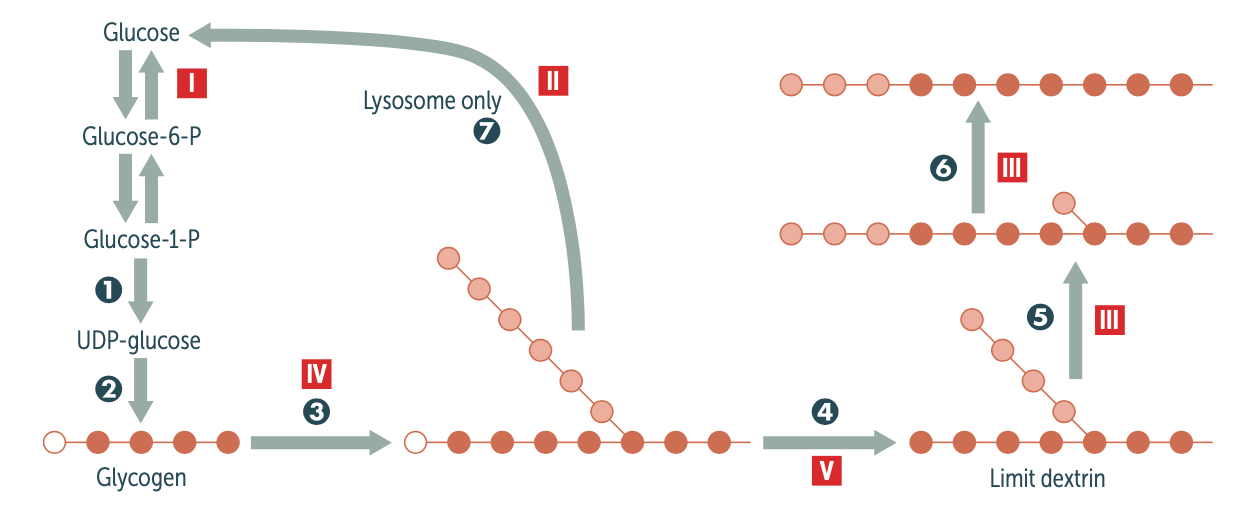
What type of GSD is caused by a defect in the glucose-6-phophatase enzyme, what is the role of this enzyme and what tissues is it most commonly expressed in? how common is this type of GSD?
Type I (Von Gierke disease) and is common in about 25% of GSD cases.
Glucose-6-phosphatase allows for the hydrolysis of glucose-6-phosphate to glucose and inorganic phosphate during gluconeogenesis which is primarily carried out in the liver. This deficiency leads to an accumulation of glycogen in the liver and kidneys.
300
What is the most common mode of inheritance of GSD?
mostly autosomal recessive (types I, II, III, and V)
300
Nothing to see here. move on
Did I Stutter?...... move on PLEASE

300
Why should foods rich in fructose and galactose be avoided in patients with Von Gierke Disease?
Foods rich in fructose and galactose should be avoided in patients with GSD type I because these sugars must first be converted to glucose-6-phosphate before they can be utilized in the body. in VGD there is a deficiency in Glucose-6-phosphatase, therefore those sugars would not be properly broken down.
400
What cancer is most seen in GSD?
Adenomas are occasionally seen and can progress to hepatocellular carcinoma
400
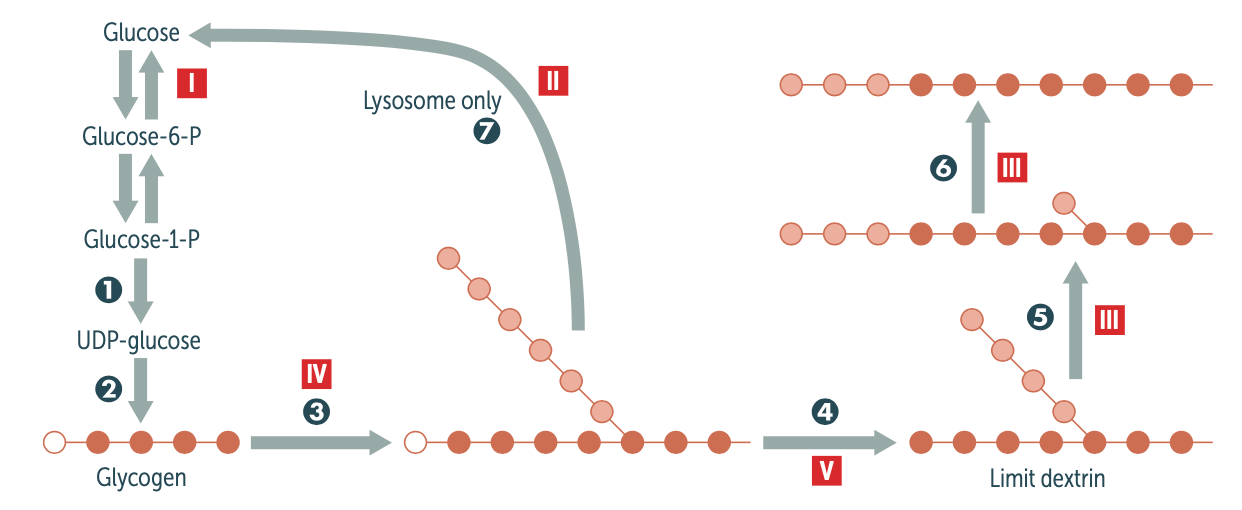
Explain the biochemical pathway in glycogen metabolism involved/affected in McArdle disease (type V). be sure to mention the enzyme affected
Glycogen is stored and undergoes glycogenolysis to maintain blood sugar at appropriate levels. Glycogen phosphorylase liberates glucose-1-phosphate residues off branched glycogen until 4 glucose units remain on a branch. Then 4-α-d-glucanotransferase (debranching enzyme ) moves 3 of the 4 glucose units from the branch to the linear linkage. Then α-1,6-glucosidase (debranching enzyme ) cleaves off the last residue, liberating a free glucose.
400
What is the most common age of onset of GSD?
Presentation during infancy or childhood however, McArdle disease (GSD type V) occurs among adults. In addition to a classic infantile form, Pompe disease (GSD type II) also has a late-onsetform which occurs in adults. Types I and II may also present early during the neonatal period.
400
What are some limitations of using histology to diagnose GSD?
The lack of morphological specificity implies that a complete enzyme analysis be performed on each biopsy for GSD.
400
What are some general therapies involved in management of GSD?
- Most forms of GSD can be managed effectively with dietary therapy (e.g., uncooked corn starch, glucose preparations) with the aim of preventing hypoglycemia and/or muscle symptoms
500
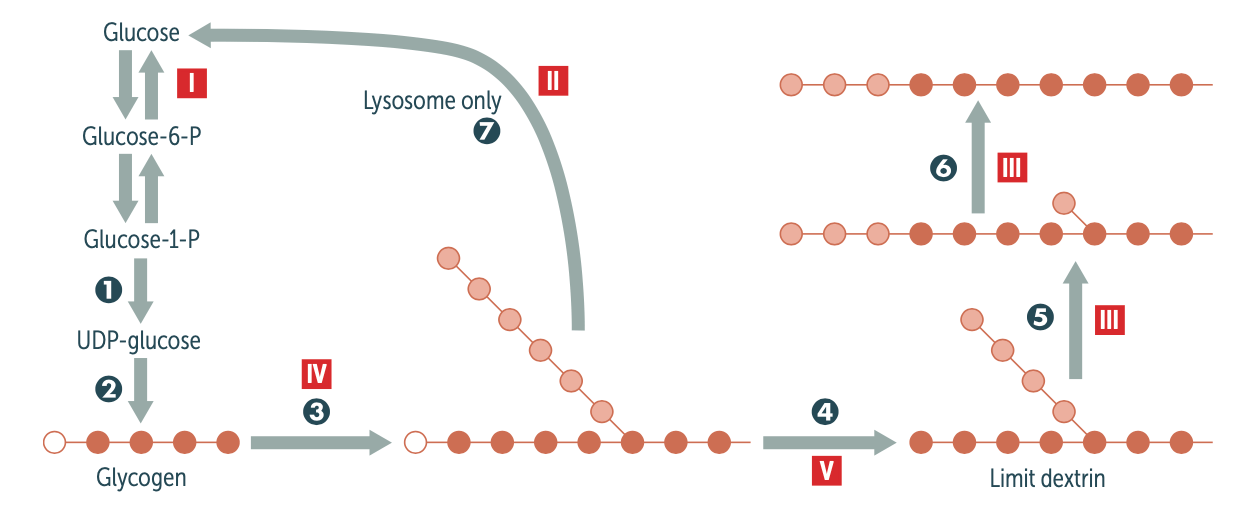
which Type on GSD is characterized by exercise intolerance and cardiomyopathy and eventually early death? What enzyme is affected in this type and how common is this type?
- Type II (Pompe disease), seen in 15% of patients
- Lysosomal acid α-1,4- glucosidase (acid maltase). A small amount of glycogen is degraded in lysosomes by α-1,4-glucosidase (acid maltase).
500
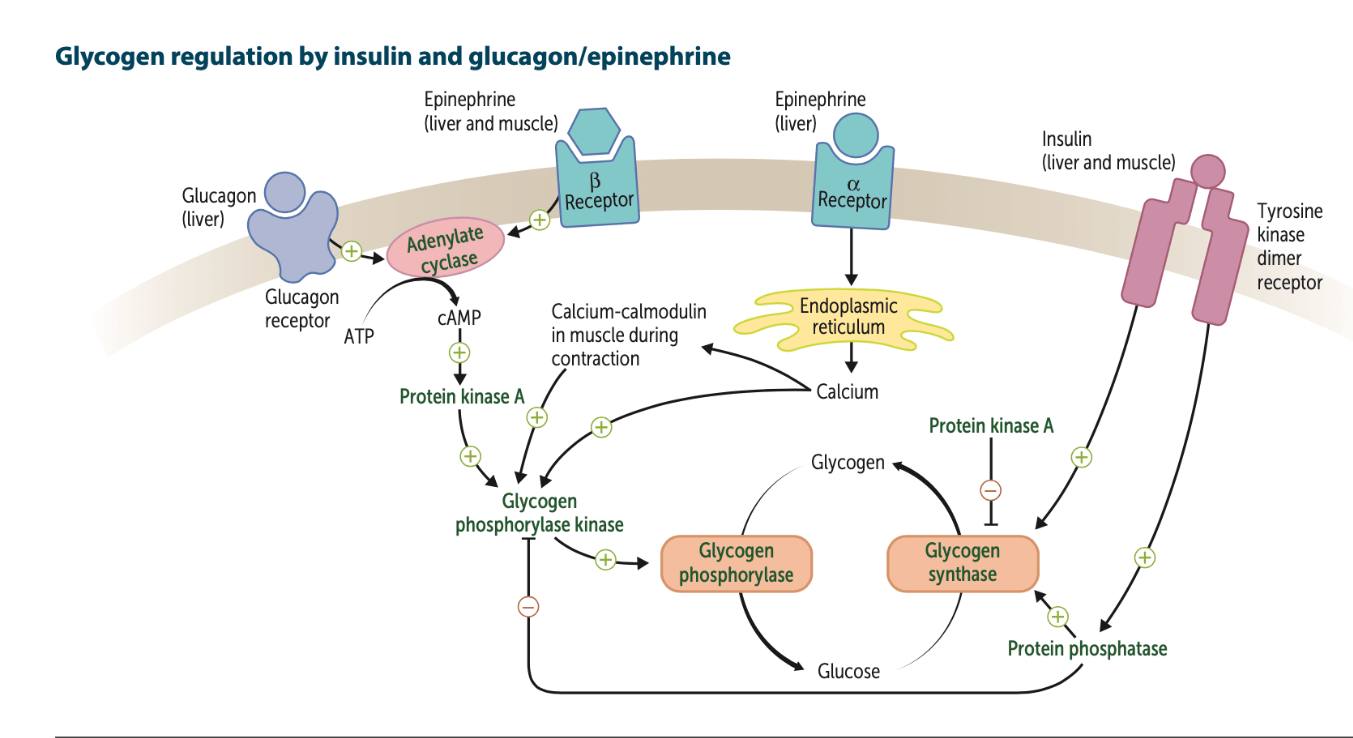
What hormones control/regulate glucose metabolism? explain the signaling pathway involved in their regulation of glycogen. make sure to mention the dominant tissue type used for each hormone.
The metabolism of glucose is primarily controlled by hormones such as insulin and glucagon.
- The insulin transduction pathway is a biochemical pathway which increases glucose storage into glycogen (promoting glycogenesis) and reduces/inhibits the breakdown of glycogen (glycogenolysis) where the rate limiting enzyme is glycogen synthase
- Glucagon is released in fasting states and promotes glycogenolysis where the rate limiting enzyme is glycogen phosphorylase, while inhibiting glycogenesis
These hormones are therefore involved in maintaining glucose homeostasis. These pathways are also influenced by fed versus fasting states, stress levels, and a variety of other hormones (cortisol, epinephrine, etc.)
500
Explain the importance of newborn screenings in GSD
Newborn screening enables the early detection of metabolic diseases and early initiation of appropriate dietary restrictions helps prevent disease manifestations.
500
Describe a pertinent histological feature seen in liver biopsies of patients Type 1 GSD and a feature seen in GSD not specific to a type.
- Deficiency of glucose-6-phosphatase results in accumulation of glycogen in hepatocytes. The liver is enlarged. The hepatocytes are swollen and a mosaic histological pattern with compression of the sinusoids is seen. Macro- and/or microvesicular steatosis can be present.
- Mosaicism, the most sensitive criterion in the diagnosis of GSD, is not type-specific.
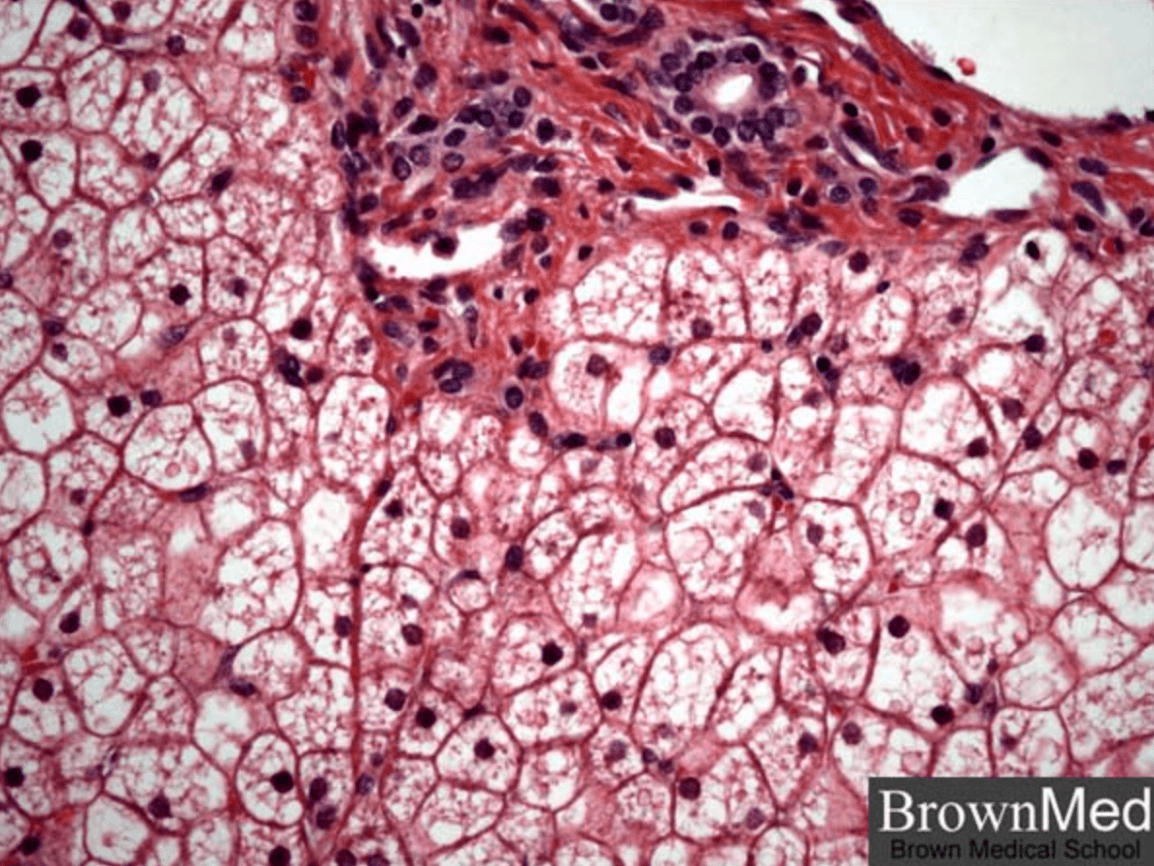
500
What is a definitive therapy for Liver GSD? what about cardiac GSD?
- Enzyme replacement therapy is available for some forms of GSD
- A liver transplant may be required in the case of liver GSD that progress to liver cirrhosis and/or result in poor metabolic control.
- Cardiac involvement
- Severe conduction defects: pacemaker implantation
- Severe cardiomyopathy: heart transplantation (see “Therapy” in “Hypertrophic cardiomyopathy”)