Anatomy
Peripheral Nerve
Myopathies
More Anatomy
Muscular Dystrophies
100
Which nerve supplies the gluteus minimus, gluteus medius and gluteus maximus?
Inferior gluteal nerve (maximus)
Superior gluteal nerve (minimus and medius)
Bonus points function of each muscle:
100
Dr. Bartnick is seeing a patient in her clinic presenting with progressive weakness and paresthesia for the past 3 weeks. The patients wants to provide a detailed hx but Dr. Bartnick has no time to waste, being chief and all. She says and I quote, "Why would i listen to your words when the journey towards the truth can be so much more fun". With a smirk she proceeds to electrocute and stab the patient with a needle AKA performs a NCS/EMG.
The test shows absent H responses & absent F waves, some of the waveforms from the study are shown below. The most likely diagnosis is...

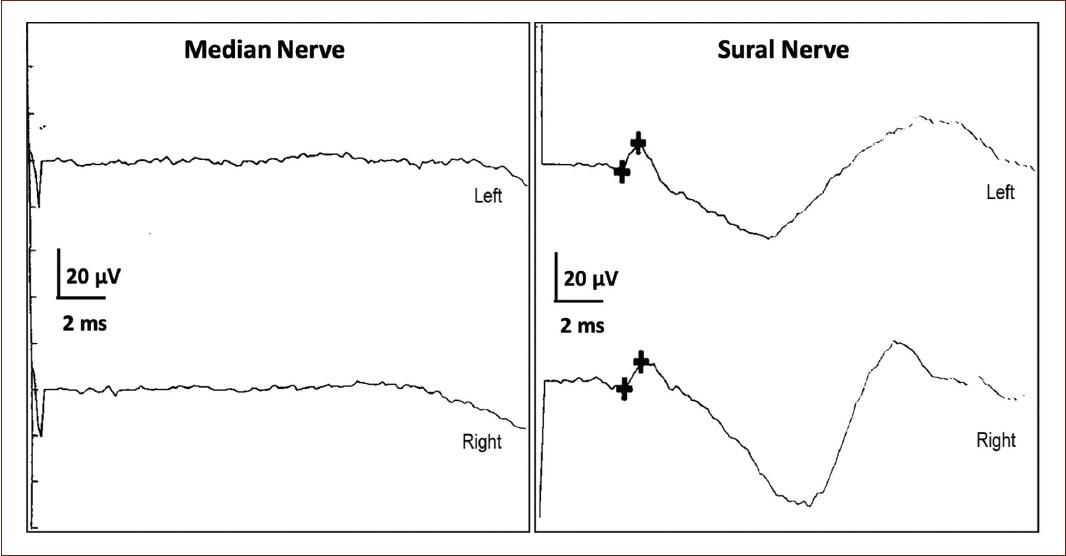
What is AIDP
Earliest sign on NCS: Delayed/Absent or impersistent F waves or H reflexes (reflecting proximal demyelination/ note AIDP starts at root level as polyradiculopathy).
Motor NCS show prolonged distal latencies and conduction block or temporal dispersion. (50% have these at week 2 and 85% by week 3)
Lae findings: Conduction velocities on MCS become slow. Sensory studies (classically) show a so called "sural sparing pattern" i.e the upper extremity nerves have absent or reduced SNAPs but sural SNAP is normal (AIDP preferentially affects small diameter myelinated fibers)
Reduced/absent SNAPs in the UE with normal sural SNAPs (sural sparing) highly specific for diagnosis of AIDP
100
An AD disorder due to a defect in the ryanodine receptor-1 gene (RYR1) on chromosome 19q, presents with neonatal hypotonia and weakness with skeletal abnormalities such as hip dislocation and clubfoot.
Central Core Disease, of note pathology shows cores in type 1 fibers on histology-areas lack mitochondria and oxidative enzymes on histology.
100
Which two nerves originate proximal to the brachial plexus and which muscles to they innervate?
1.Dorsal scapular-innervates the levator scapulae and rhomboids.
2. Long thoracic nerve-innervates the serratus anterior.
100
Discuss the genetic mutation in the dystrophin gene on Xp and its two phenotypic disorders.
1. Duchenne’s MD- out-of-frame mutation (on muscle biopsies stained with anti-dystrophin antibodies there is NO dystrophin) vs. 2. Becker MD have in-frame mutations, muscle biopsies show REDUCED dystrophin.
200
Discuss the difference in clinical presentation of a L5 radiculopathy versus a common peroneal neuropathy.
Clinical findings can aid in determining the etiology of a patient’s condition. L5 radiculopathy and peroneal neuropathy can both present with weakness of the foot dorsiflexors and toe extensors.
L5 radiculopathy may present with weakness during foot INVERSION whereas peroneal neuropathy will not.
Additionally, reflex changes at the patella, medial hamstring and Achilles tendon can distinguish a L4, L5 or S1 radiculopathy from a common peroneal neuropathy.
Sensory changes to light touch or pinprick may not improve the clinical picture as dermatomal patterns and peripheral nerve distributions can have much overlap and sensory evaluation may be prone to subjective bias.
Finally, adverse nerve root tension, including femoral nerve stress test and straight leg raise can indicate a lumbar nerve root involvement which is absent during peroneal neuropathy. On the other hand, passive or forceful ankle inversion tensions the peroneal nerve which may reproduce symptoms of a peroneal neuropathy.
200
Name the gene mutation for each of the following forms of Charcot Marie Tooth disease and the mode of inheritance
- CMT1a
- HNPP
- CMT1X:
Bonus points if you know this:
CMT2A: mode of inheritance, type of neuropathy
CMT4: mode of inheritance
Autosomal dominant: CMT1 and CMT2
Autosomal recessive: CMT 4
X linked CMTX
CMT1-most common type of CMT (40-50%) (6 subtypes) all are demyelinating hereditary motor and sensory neuropathy with nerve biopsy findings of demyelination and onion bulb formation.
*CMT1A: MC CMT overall. 70-80% of CMT1 cases. Associated with a duplication on PMP22 gene on 17p.
* CMT1b MPZ gene on chromosome 1 (more severe, 10% of CMT1 cases)
* HNPP: AD. Deletion in PMP22 gene on chromosome 17p. Clinically present w/ recurrent episodic painless mononeuropathies in adolescence or young adults (variable)
CMT2-axonal form in which motor CVs are normal or near normal and nerve biopsy reveals axonal loss without prominent demyelination. AD inheritance begins later in life 2-3rd decades, foot/spinal deformities are less prominent-similar clinical features except peripheral nerves are not enlarged and upper extremity involvement, tremor and general arreflexia occur less frequently.
CMT4 Autosomal recessive
200
A mutation on the 1q gene, that encodes the alpha-1 subunit of L-type calcium channels results in this autosomal dominant condition. Discuss exacerbating factors and treatment.
Hypokalemic Periodic Paralysis-characterized by episodes of weakness that last for hours or days, weakness triggered by a large carbohydrate load or exercise, treated with rest/fasting.
200
Describe the difference in injury resulting in a medial versus lateral winging of the scapula?
The serratus anterior innervated by the long thoracic causes medial winging. The trapezius innervated by the spinal accessory nerve causes lateral winging.
200
This muscular dystrophy (has three forms of inheritance) and is associated with early joint contractures, weakness in humero-peroneal distribution and cardiac abnormalities.
Emery-Dreifus Muscular Dystrophy.
300
Discuss the stretch reflex.
Hitting a tendon with a reflex hammer causes a stretch to be applied to a muscle, which is detected by the muscle spindles. Due to the stimulation of the muscle spindles an excitatory impulse is sent through Ia afferent fibers (sensory nerve) to the alpha motor neuron in the spinal cord which then fires and causes contraction of the muscle (a monosynaptic pathway).
300
What disease is associated with mutation in the SPTLC1 gene encoding a subunit of serine palmitoyltransferase and is the most common hereditary sensory neuropathy?
Hereditary Sensory and Autonomic Neuropathy Type 1.
300
Discuss the difference in pathophysiology between dermatomyositis and polymyositis and clinical presentations of both.
Dermatomyositis is a humorally-mediated microangiopathy (often present with underlying malignancy) presents with limb-girdle weakness and characteristic heliotrope rash (perio-orbitally) and seen on the knuckles (Gottron’s papules). Associated with EKG abnormalities, interstitial lung disease (anti-synthetase antibody) and perfasicular atrophy on pathology. Polymyositis is T-cell mediated process presenting with limb-girdle distribution weakness. CK is often elevated and muscle biopsy shows endomysial inflammation-diagnosis requires the presence of CD8+ T cells in major histocompatibility complex class 1 positive muscle fibers.
300
What syndrome is characterized by pain in the forearm, weakness of forearm pronation, weakness with thumb flexion and the terminal phalanges of the second and third digits without loss of sensation?
AIN (anterior interosseous syndrome)
300
What is the name of the disorder with AD inheritance due to a deletion of D4Z4 repeats on chromosome 4q?
Fascioscapulohumeral Muscular Dystrophy- associated with asymmetric weakness involving the face, muscles that stabilize the scapula, proximal upper extremities and dorsiflexors of the foot.
400
Which muscle of the lower extremity is the only muscle innervated by the peroneal nerve above the fibular neck?
Biceps femoris-short head.
400
Name three variants of AIDP.
1. AMSAN (acute sensory and motor neuropathy) fulminant axonal variant-patients develop an acute and rapidly progressive course often with profound quadriparesis and severe muscle wasting and requiring ventilator support, EDX markedly reduced or absent CMAPS without significant conduction slowing.
2. AMAN (acute motor axonal neuropathy)-presents with acute flaccid paralysis without clinical or electrophysiological involvement of sensory nerves, EDX with prominent axonal features (reduced CMAP amplitudes but normal motor distal latencies and conduction velocities).
3. MFS (5% of GBS cases)-triad of ophthalmoplegia, ataxia and areflexia-associated with serum IgG antibodies to the ganglioside GQ1b.
400
Name the most common inflammatory muscle disease in patients over 50 years old, involves the quadriceps and wrist and finger flexors. Discuss EMG findings (typical findings in myopathic processes).
Inclusion body myositis. EMG findings: Increased insertional activity, low amplitude short duration motor unit action potentials, early recruitment of motor unit action potentials. Pathology-light microscopy shows- endomysial inflammation, eosinophilic cytoplasmic inclusions, rimmed vacuoles, amyloid deposition.
400
A lesion of which nerve results in finger drop with weakness in wrist extension without sensory involvement?
Posterior Interosseous nerve (deep branch of the radial nerve).
400
What disorder displays AD inheritance due to an amplified CTG repeat in the DMPK gene on chr 19q; associated multi-organ involvement to include cardiomyopathy and conduction defects, insulin resistance, hypogonadism, hypoventilation? Discuss its analog and the differences between the two.
Myotonic Dystrophy Type 1. Its analog DM2- aka proximal myotonic myopathy (PROMM)-unlike DM1 involves proximal muscles, less cardio/pulmonary involvement, no congenital form , associated with less atrophy and due to a tetranucleotide repeat (CCTG repeat expansion in the ZNF9 (zinc finger protein 9) at 3q.
500
Discuss Type I, Type IIb, Type IIa muscle fibers including which enzymes and form of metabolism they use and where/when they are commonly found and used
Type I-slow twitch contain oxidative enzymes and use aerobic metabolism, produce steady force and are fatigue resistant. Often found in truncal/postural muscles.
Type IIb muscle fibers-fast fatigable contain glycolytic enzymes and use anaerobic metabolism, produce greatest amount of force but are easily fatigable.
Type IIa fibers-fast, resistant fibers, contain both oxidative and glycolytic enzymes and are resistant to fatigue. Type II fibers tend to be found in the extremities.
500
Name the diseases associated with the following:
1) Inherited deficiency of the lysosomal enzyme galactocerebroside B-galactosidase, characterized by extensive CNS/PNS demyelination and the presence of multi-nucleated macrophages in the cerebral white patter? 2) The gene defect chromosome 10 encoding the peroxisomal enzyme phytanoyl-CoA-hydroxylase, cardinal manifestations include pigmentary retinal degeneration, chronic hypertrophic neuropathy, ataxia, and other cerebellar signs (nystagmus and intention tremor).
3) AR disorder due to mutations in the gene that encodes MTP (microsomal triglyceride transfer protein) on chromosome 4, clinical present with ataxia, neuropathy, retinitis pigmentosa, acanthocytosis.
4) AR disorder characterized by severe deficiency of plasma alpha or high-density lipoproteins resulting in deposition of cholesterol esters in many tissues including the reticuloendothelial system and peripheral nerves.
5) AR disorder of sulfatide metabolism caused by deficiency of the lysosomal enzyme, arylsulfatase A and subsequent accumulation of sulfatides in brain, peripheral nerves and other tissues.
1. Globoid cell leukodystrophy (Krabbe disease).
2. Refsum’s disease.
3. Abetalipoproteinemia/ Bassen-Kornweig syndrome.
4. Tangier disease.
5. Metachromatic Leukodystrophy.
500
This myopathy is this most common metabolic cause of recurrent myoglobinuria.
Carnitine Palmitoyl Transferase-II Deficiency-defect on ch 1-typically presents in adolescence. Patients present with tenderness and swelling of muscles with sustained aerobic exercise (or other stressors such as infection)-associated with increased CK and myoglobinuria, patients are normal between episodes.
500
Draw the brachial plexus: labeling roots/trunks/divisions/cords/divisions.
Go to the board! YAY!
500
Which muscular dystrophy is AD-due to a GCC repeat expansion in the gene encoding PAPB2 (poly-A binding protein-2) on 14q?
Oculopharyngeal muscular dystrophy presents in middle age with asymmetric ptosis due to asymmetric involvement of the levator palpebrae muscles; pharyngeal involvement results in dysphagia and dysarthria.