MNDs
Muscular Dystrophies
Inflammatory Muscle Disease
Metabolic Myopathies
Mystery Category: what's in a name?
100
Amyotrophic lateral sclerosis is a mix of damage to these to neuron populations.
What are the upper and lower motor neurons?
100
This X-linked genetic muscle disorder is caused by defects in the largest gene in the human genome, at ~2,200,000 million bases and accounting for 1/1000th of the total genome.
What is Duchenne/Becker Muscular Dystrophy (caused by mutations in the DMD gene for Dystrophin)?
100
A patient walks into your clinic with weakness and rashes, including the specific type seen here associated with this type of muscle disorder.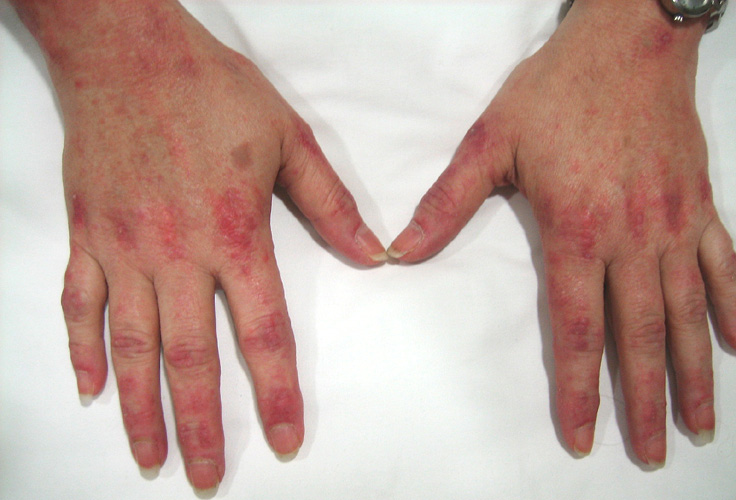
What are Gottron's papules associated with dermatomyositis?
100
This disorder may represent with CNS and PNS findings, including epilepsy, ataxia and jerking, optic atrophy, polyneuropathy, hearing loss, migraine, and myopathy, with muscle biopsy seen below.
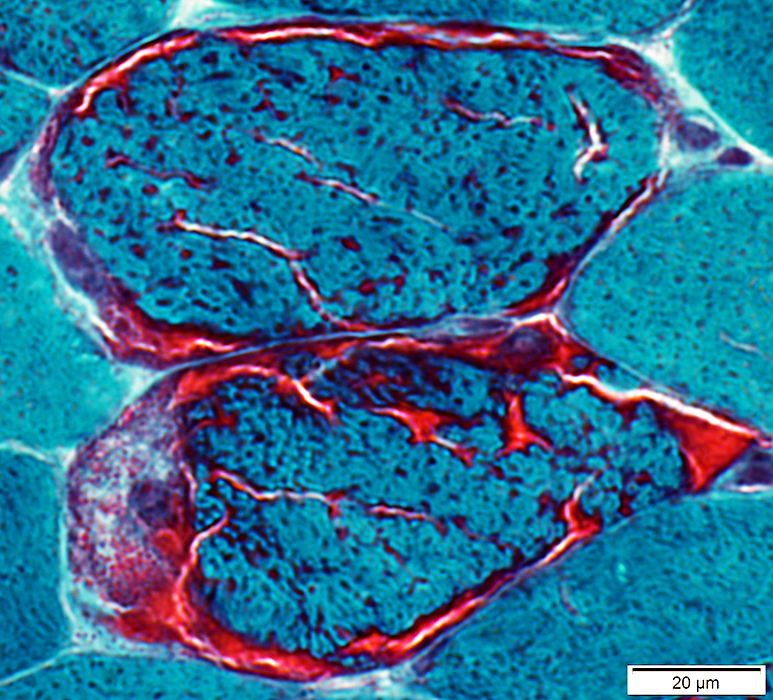
What is MERFF, most commonly caused by mtRNALys A8344G?
Disruption of mitochondrial function can lead to numerous CNS and PNS pathologies including CPEO, MERFF, MELAS, MNGIE, Alpers, Kearns-Sayre, Leigh, NARP, etc. The ragged red fibers on Gomori Trichrome are highly testable and easy to get if recognized.
100
Milton S. Hershey's middle name.
What is Snavely?
200
These are also called Pyramidal Betz Cells.
What are upper motor neurons in Layer V of the primary motor cortex?
200
This facial pattern is associated with this disease, caused by this mutation pattern, in this gene.
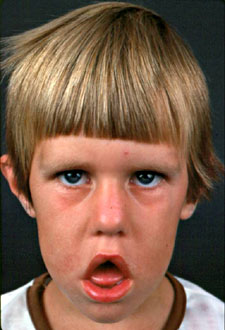
What is Myotonic Dystrophy?
Myotonic Dystrophy, caused by CTG trinucleotide expansions in the DMPK gene, is known to cause "hypotonic facies" with drooping lower face, eyelid ptosis, and muscle wasting (the "hatchet facies") along with male-pattern baldness.
200
Though not a primary autoimmune muscle disorder, this early-presenting myopathy may have prominent inflammatory infiltrates in addition to the other dystrophic findings seen here and can be treated with steroids to prolong the ambulatory period and slow cardiomyopathy.
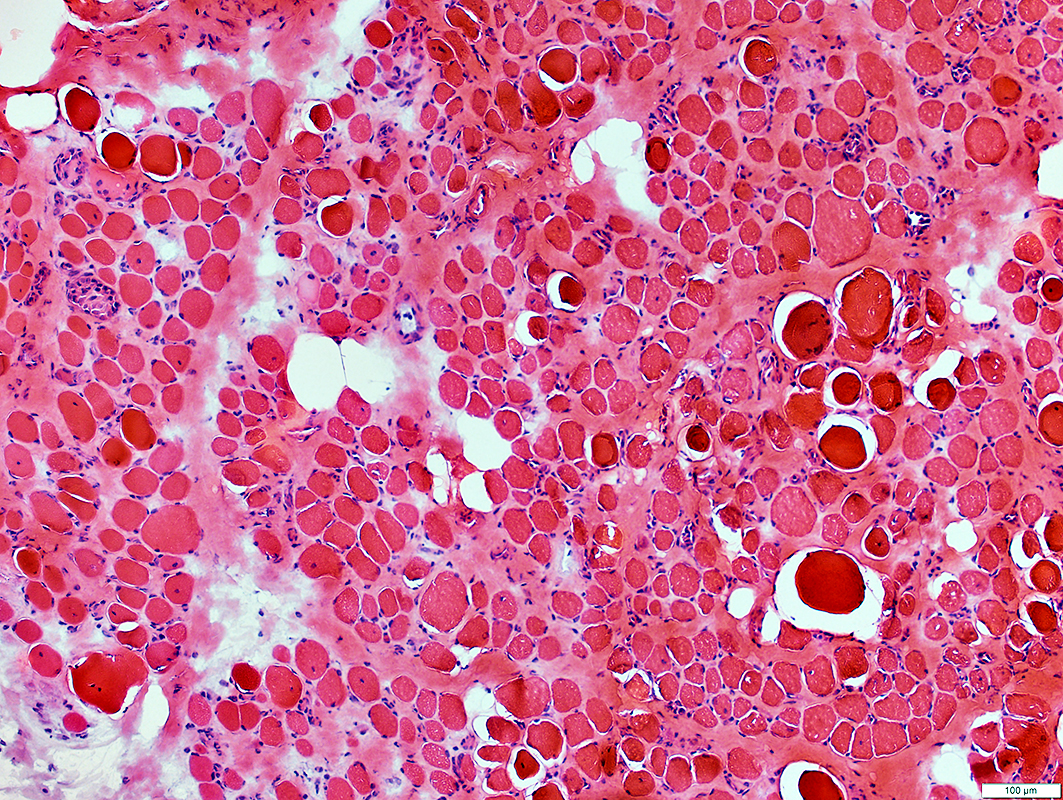
What is Duchenne Muscular Dystrophy?
In addition to more novel treatments like exon-skippers and micro-dystrophin gene therapies, steroids have been a mainstay in prolonging the ability to walk and may have an effect on heart function as well. A good amount of the challenge in patients with DMD was mitigating steroid effects.
200
If a patient presented to your clinic with muscle weakness, elevated CK, prominent orthopnea and axial atrophy with lordosis with bilateral scapular winging, vacuolar changes on muscle biopsy, and strong family history of cardiomyopathy, this is the gene you would test first.
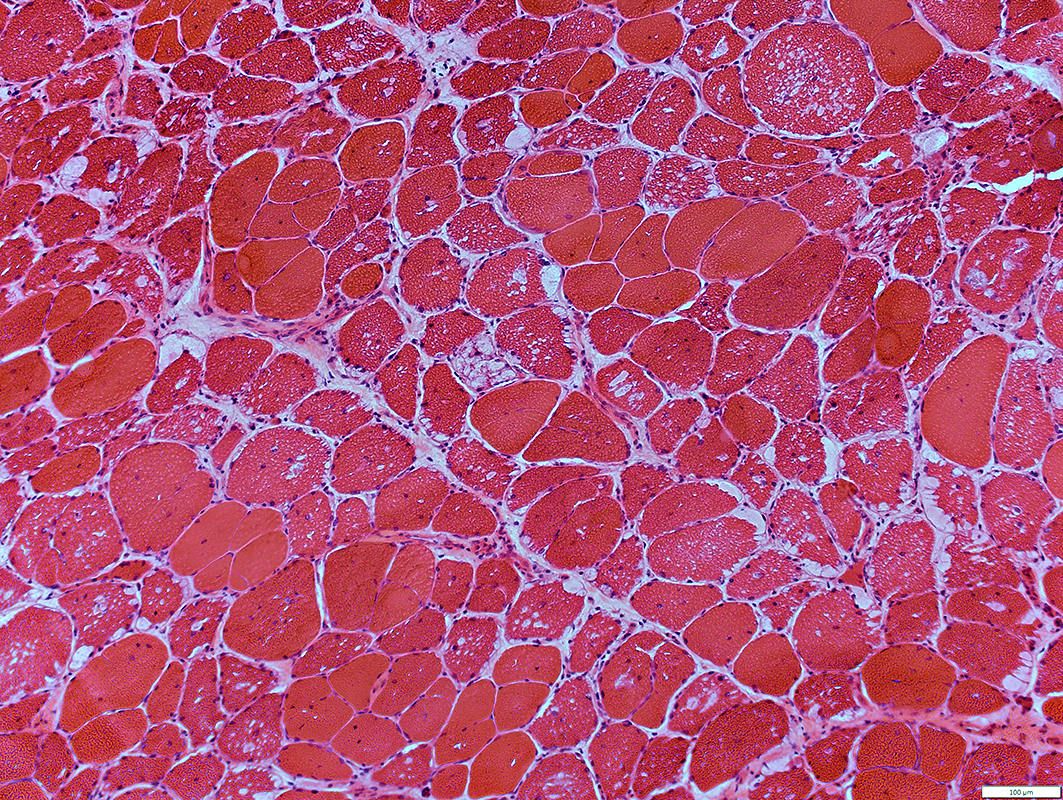
What is GAA and Pompe's Disease?
200
This is James Grogan's daughter's name.
Who is Eleanor Ruth Grogan?
300
This motor neuron disorder has exclusively upper motor neuron pathology and UMN signs on exam.
What is primary lateral sclerosis?
(HSP would be an alternative answer)
300
A patient presenting with mutations in this gene, the most common cause of Limb-Girdle Muscular Dystrophies, may present with pure proximal weakness, asymptomatic hyperCKemia, or Erb's Dystrophy (pictured below, periscapular wasting>>pelvic), and notably has no cardiac involvement.
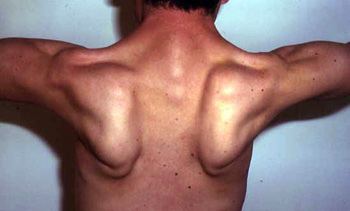
Calpain-3 (CAPN3 gene) is a calcium-activated non-lysosomal cysteine thiol-protease that cleaves proteins into polypeptide chains and associates with several muscle proteins including:
- C-terminus of titin (connectin) in muscle
- Filamin C
- Calmodulin kinase IIβ (CaMKIIβ)
- Cardiac ankyrin repeat protein (CARP; ANKRD1)
- Dysferlin
- Myospryn (CMYA5)
- AHNAK
CAPN3 mutations cause approximately one third of LGMD cases worldwide, can present with many different manifestations, and is one of the only ones without prominent cardiomyopathy.
300
These structural changes are seen in what muscle disorder (black arrow affected, white arrow unaffected)
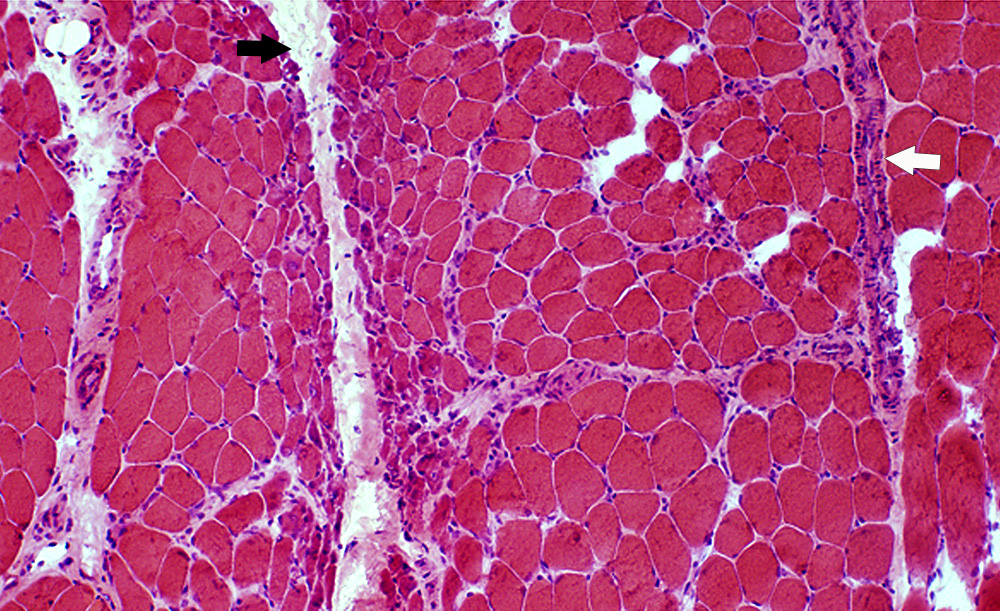
What is perifascicular atrophy associated with dermatomyositis?
Immune-mediated microangiopathic changes in the perimysial blood vessels causes atrophy in the avascular more peripheral fibers, leading to the classic appearance of smaller fibers on the edges of fascicles. May be seen more with Tif1-γ and NXP2 mediated disease.
300
Just kidding, more muscular dystrophies!
Myopathies associated with this protein (pictured below) can be associated with LGMD2B, is one of the most common causes of distal muscular dystrophy (e.g. Miyoshi type), and has prominent cardiac involvement.

What is dysferlin, encoded by the DYS gene?
Dysferlin is one of many proteins involved in muscle excitation-contraction coupling, including those in the costamere and anchoring and regulatory proteins.
300
This is the name of the physician who first characterized ALS.
Who is Jean-Martin Charcot?
400
These two genes account for nearly half of familial ALS and include quirks such as ultra-rapid progression and high FTD risk.
What are SOD1 and C9orf72?
400
This discharge on EMG needle examination can be seen in what disorders?
What are Myotonic Dystrophy Type 1, Proximal Myotonic Myopathy (DM2), myotonia congenita, paramyotonia congenital, myotonia permanens, myotonia fluctuans, IMNM, etc.
400
In a patient with this muscle disorder, they may present with:
- rapidly progressing weakness and muscle pain
- brief myotonic discharges on EMG needle examination
- have a stark lack of inflammatory infiltrates on pathology
- and can be triggered by multiple medications and rarely Pu-erh tea.
What is Immune Mediated Necrotizing Myopathy (or Necrotizing Autoimmune Myopathy)?
IMNM is a rapidly progressing necrotizing myopathy most commonly caused by antibodies to SRP or HMG-CoA-R. These require prompt recognition and treatment due to muscle fiber necrosis and risk of irreversible damage. They often require longterm management with IVIG, steroids, and B-cell depletion (CD20 MABs). Statins and other HMG-CoA inhibitors including pu-erh tea and red yeast rice.
400
A 10 year old with post-exercise muscle cramps and weakness comes in to your clinic. While experiencing a cramp during an EMG, you notice electrical silence?

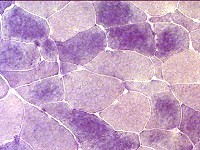
These are the findings on his muscle biopsy (with normal phosphorylase stain on right), and his symptoms are associated with this muscle disorder.
What is McArdle's Disease (GSDV) with myophosphorylase deficiency and absence on muscle biopsy phosphorylase immunohistochemistry?
400
This is the name of one of Dr. Grogan's rabbits!
Who are Cirrus, Kiwi, or Juniper?
500
This is the mechanism by which nusinersen (Spinraza) modifies that gene's expression in Spinal Muscular Atrophy.
What is an antisense oligonucleotide that causes post-transcriptional modifications of SMN2 leading to preferential formation of SMN protein and motor neuron rescue?

Nusinersen binds to splice-silencing-site in SMN2 intron 7, inhibiting action of other splice-factors and promoting the full SMN protein lost due to SMN1 gene deletions. It is intrathecal Q4M after loading doses. Other options include oral risdaplam and gene therapy with Onasemnogene Abeparvovec-xio (Zolgensma).
500
Patients with a mutation in this gene affecting this ion channel may have a presentation of proximal and distal muscle weakness with contracture, pure distal myopathy, isolated cardiomyopathy, benign hyperCKemia, or a rare rippling muscle disease seen here.

What is the caveolin-3 Ca channel encoded by the CAV3 gene?
500
This muscle disease, though not primarily autoimmune, is known to have mononuclear inflammatory infiltrates as well as TDP-43 and other protein deposition and these particular vacuolar findings on pathology.
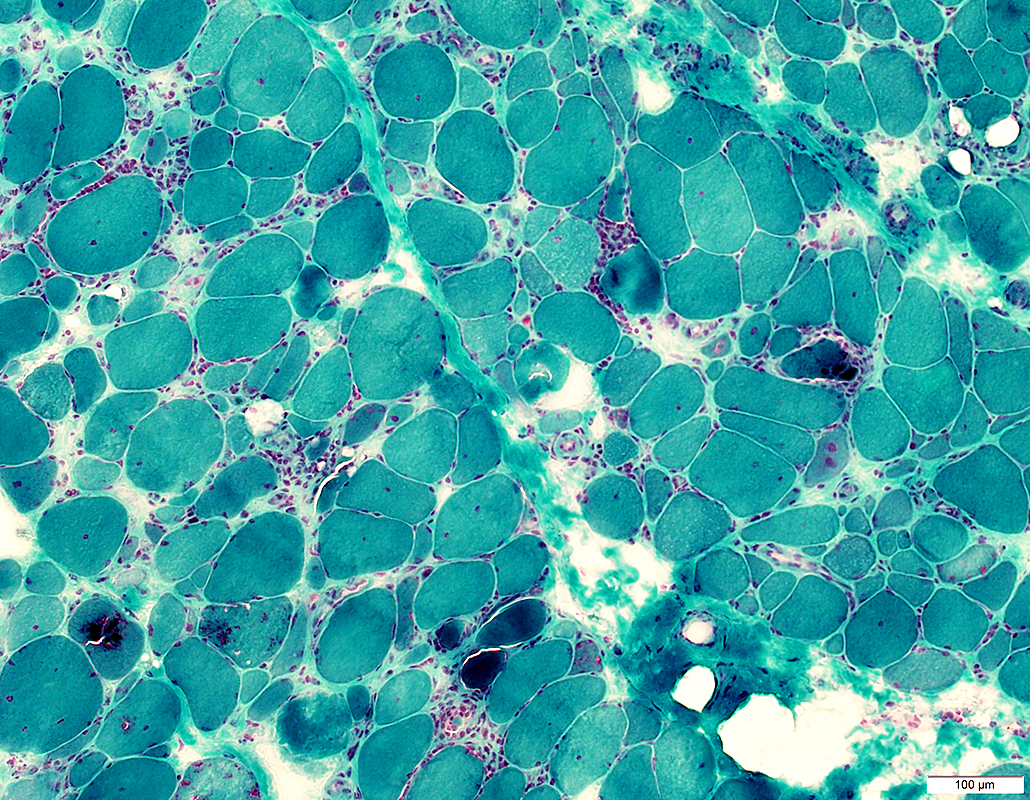
What are inclusion bodies and rimmed vacuoles in sporadic inclusion body myositis?
sIBM is the most common acquired myopathy after age 50, disproportionately affecting finger flexors, knee extensors, and swallowing muscles.
500
This metabolic myopathy most commonly manifests as abrupt, sudden, recurrent rhabdomyolysis often in the setting of acute illness and fasting when the body cannot process this macronutrient. After resuming normal nutrition, they usually return to baseline though can worsen over time if repeatedly triggered.
What are disorders of fatty acid oxidation, most commonly caused by mutations in CPT2?
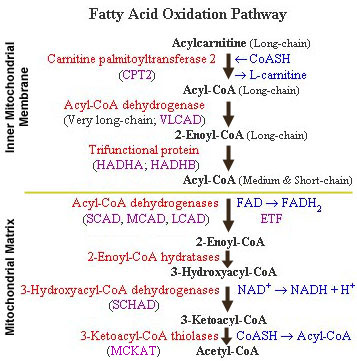
500
This is the name of James Grogan's favorite resident.
What even is this?
He loves all of you equally (come get your candy). Next question.